#代码示例-----
# Installing necessary packages
if (!requireNamespace("readr", quietly = TRUE)) {
install.packages("readr")
}
if (!requireNamespace("ggplot2", quietly = TRUE)) {
install.packages("ggplot2")
}
if (!requireNamespace("tidyverse", quietly = TRUE)) {
install.packages("tidyverse")
}
# Loading the libraries
library(readr)
library(ggplot2)
library(tidyverse)Contribution_guidance
对于可视化教程的示例指导
Example
【Figure. 示例图】
展示以上示例图的标题/图注,并对示例图xy轴或其他标识物的含义解读。
环境配置
系统要求 跨平台(Linux/MacOS/Windows)
编程语言: R
依赖资源: (填充可视化教程依赖的R包或其他资源)
数据准备
- 需包含R内置的数据集(如iris、penguins)和生物医学相关数据集(如组学数据、生存信息、临床指标等)。
- 生物医学相关数据集需上传至Bizard腾讯云以便获取插入教程的链接,来自于公共数据集的数据最佳,若由个人/组织提供需确保数据能够被公开。数据集大小需小于1MB。
# 数据读入和处理代码可以自由选择是否展示------
# 读取 TSV 数据
data <- readr::read_tsv("https://bizard-1301043367.cos.ap-guangzhou.myqcloud.com/TCGA-LIHC.htseq_counts.tsv.gz")
# 筛选并重塑第一个基因 TSPAN6 的数据(Ensembl ID: ENSG00000000003.13)
data1 <- data %>%
filter(Ensembl_ID == "ENSG00000000003.13") %>%
pivot_longer(
cols = -Ensembl_ID,
names_to = "sample",
values_to = "expression"
) %>%
mutate(var = "var1") # 添加一列来区分变量
# 筛选和重塑第二个基因 SCYL3 的数据 (Ensembl ID: ENSG00000000457.12)
data2 <- data %>%
filter(Ensembl_ID == "ENSG00000000457.12") %>%
pivot_longer(
cols = -Ensembl_ID,
names_to = "sample",
values_to = "expression"
) %>%
mutate(var = "var2") # 添加一列来区分变量
# 合并两个数据集
data12 <- bind_rows(data1, data2)
# 查看最终的合并数据集
head(data12)# A tibble: 6 × 4
Ensembl_ID sample expression var
<chr> <chr> <dbl> <chr>
1 ENSG00000000003.13 TCGA-DD-A4NG-01A 12.8 var1
2 ENSG00000000003.13 TCGA-G3-AAV4-01A 9.72 var1
3 ENSG00000000003.13 TCGA-2Y-A9H1-01A 11.3 var1
4 ENSG00000000003.13 TCGA-CC-A3M9-01A 11.6 var1
5 ENSG00000000003.13 TCGA-K7-AAU7-01A 11.5 var1
6 ENSG00000000003.13 TCGA-BC-A10W-01A 12.0 var1 可视化
1. 基础绘图
使用基础函数绘制图片的图注和简介。
例:@fig-BasicHist 显示了 TSPAN6基因在不同样本中的表达水平分布。
# 基础绘图代码示例-----
#| label: fig-BasicHist
#| fig-cap: "Basic Histogram"
#| out.width: "95%"
#| warning: false
# 基础柱状图
p1 <- ggplot(data1, aes(x = expression)) +
geom_histogram() +
labs(x = "Gene Expression", y = "Count")
p1`stat_bin()` using `bins = 30`. Pick better value `binwidth`.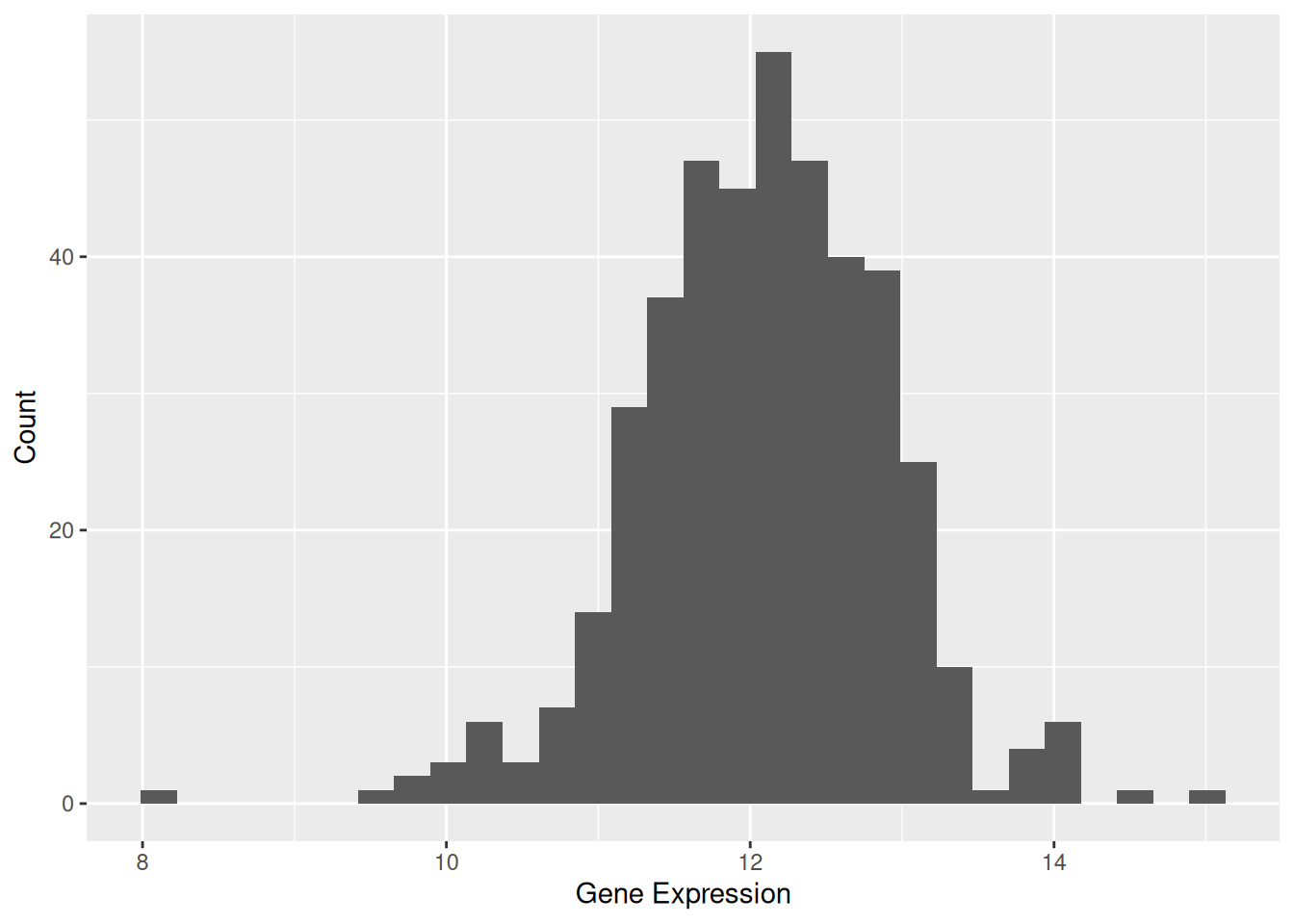
补充基础代码可以扩展的重要参数,并提供对应的绘图代码。 例: ::: callout-tip 关键参数: binwidth / bins
binwidth 或 bin参数决定了每个分区包含多少数据。修改这些值会极大地影响直方图的外观和传递的信息。 :::
# 代码示例(以补充参数bins为例)-----
#| label: fig-bins
#| fig-cap: "Key Parameters: `binwidth` / `bins`"
#| fig.width: 8
#| fig.heright: 2
#| out.width: "95%"
#| warning: false
p2_1 <- ggplot(data1, aes(x = expression)) +
geom_histogram(bins = 30, fill = "skyblue", color = "white") +
ggtitle("Bins = 30") +
labs(x = "Gene Expression", y = "Count")
p2_2 <- ggplot(data1, aes(x = expression)) +
geom_histogram(bins = 50, fill = "skyblue", color = "white") +
ggtitle("Bins = 50") +
labs(x = "Gene Expression", y = "Count")
cowplot::plot_grid(p2_1, p2_2)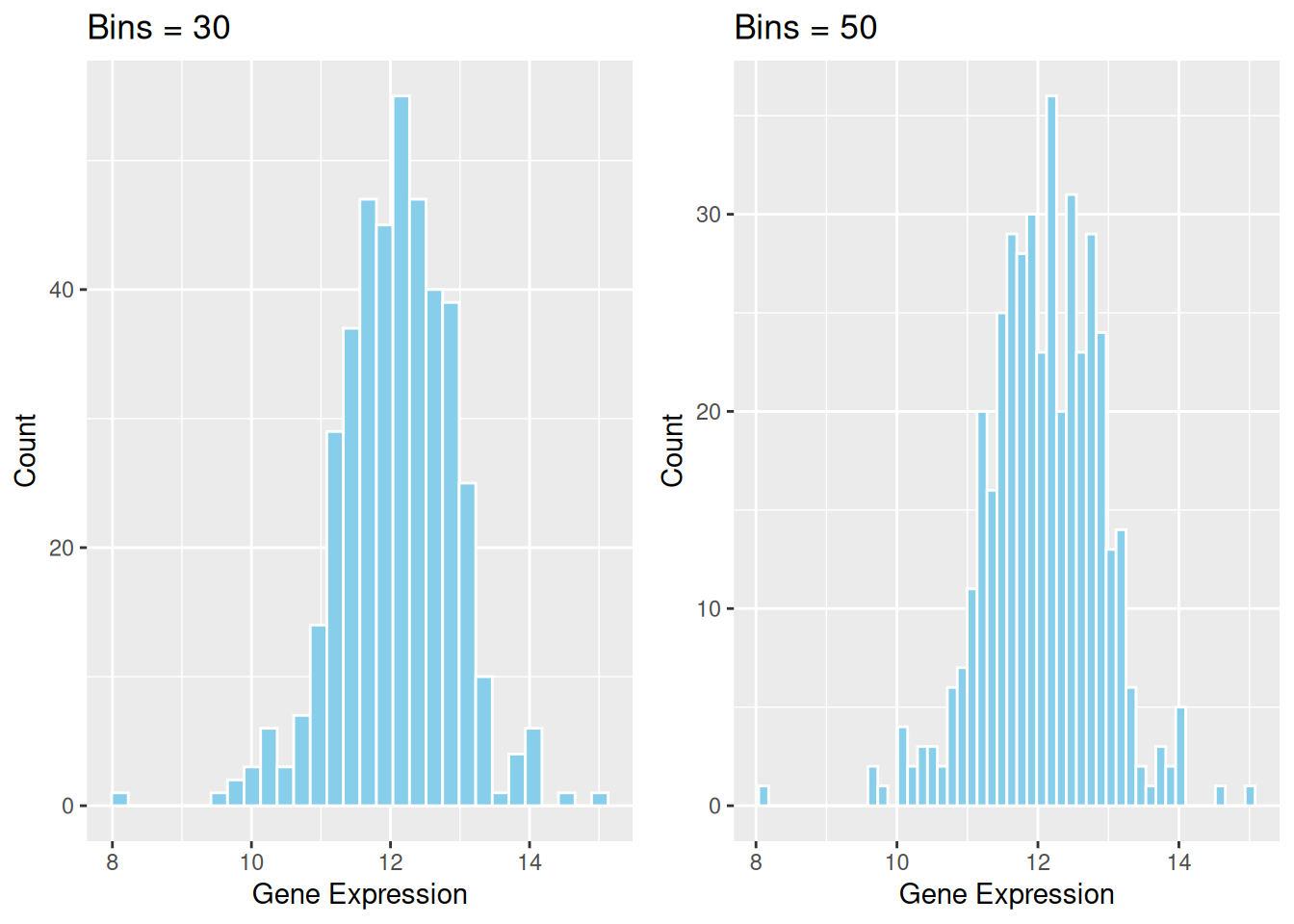
2. 更多进阶图表 (例:密度曲线和柱状图结合)
介绍复杂的可视化类型,例如使用包含更多自定义参数的函数、使用多种基础图表叠加、添加统计学检验等。 例:密度曲线可以平滑地表示数据分布。直方图依赖于分位数,而密度曲线则不同,它使用核密度估计(KDE)来平滑分布。这样就能更清楚地了解数据的整体趋势和形状。
# 进阶绘图代码示例-----
#| label: fig-DensityCurve
#| fig-cap: "Histogram with Density Curve"
#| out.width: "95%"
#| warning: false
p1 <- ggplot(data1, aes(x = expression)) +
geom_histogram(aes(y = after_stat(density)), bins = 30, fill = "skyblue", color = "white") +
geom_density(alpha = 0.2, fill = "#FF6666") +
labs(x = "Gene Expression", y = "Density")
p1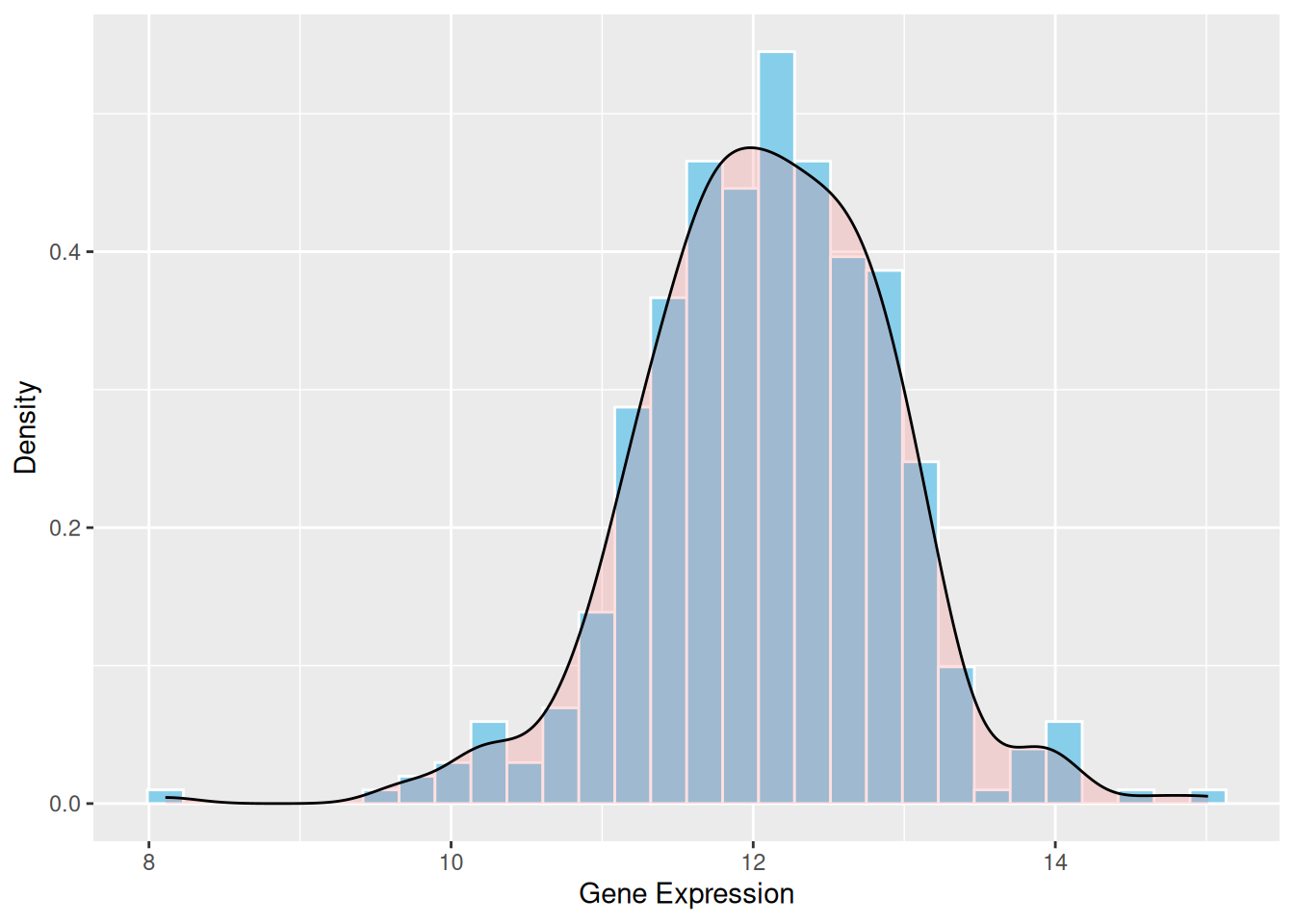
如果你有需要的话可以选择使用callout-tip添加对参数的详细描述。
应用
展示可视化图表在生物医学文献中的实际应用,如果基础图表/进阶图表被广泛应用在各类生物医学文献,则可以选择分别展示。
例: ### 1. 基础柱状图的应用
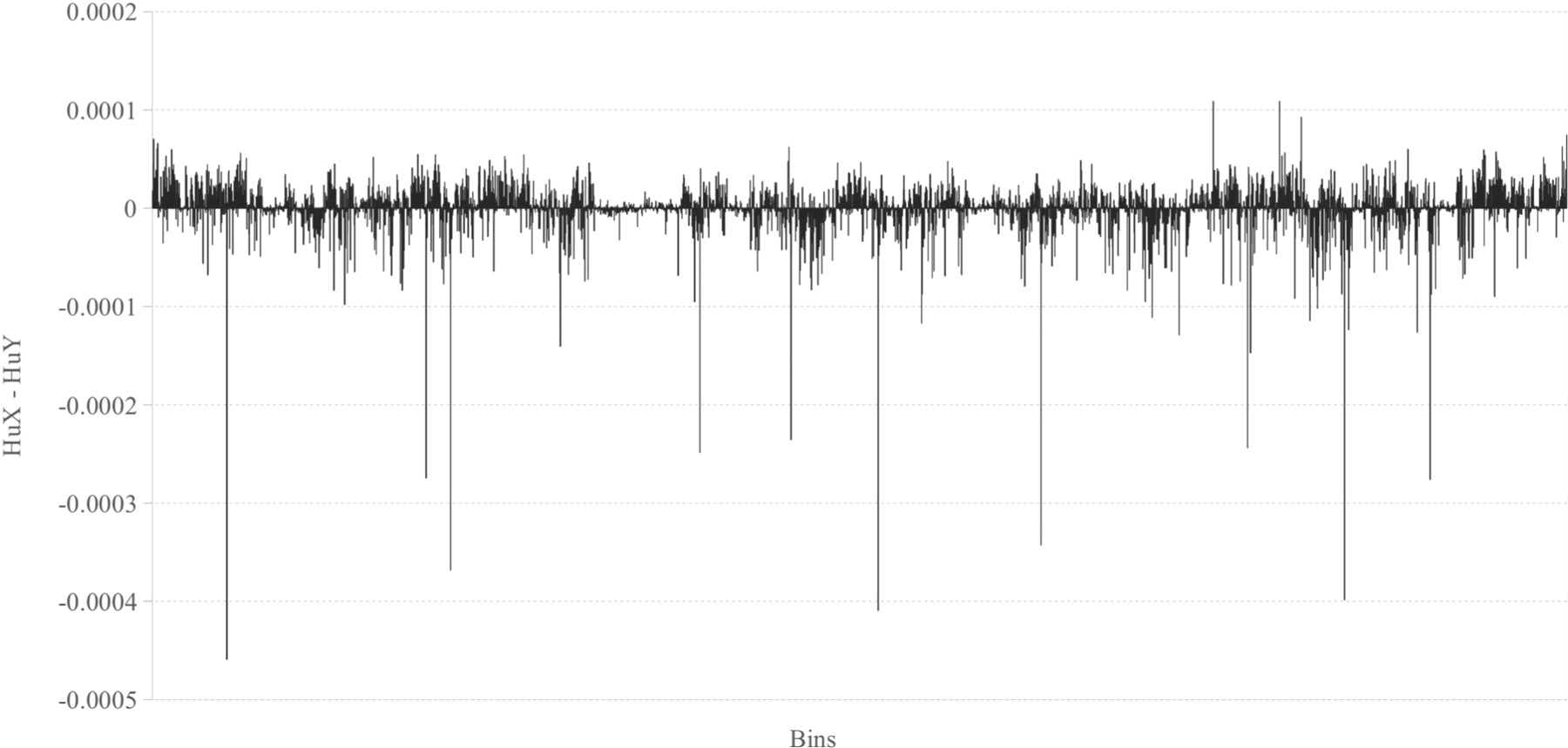
图 9 显示了 n = 6时人类X染色体和人类Y染色体直方图相对频率的差异。 [1]
需补充图片图注和来源文献信息。可视作者意愿补充对该图的代码复现。
参考文献
例: 1. Costa, A. M., Machado, J. T., & Quelhas, M. D. (2011). Histogram-based DNA analysis for the visualization of chromosome, genome, and species information. Bioinformatics, 27(9), 1207–1214. https://doi.org/10.1093/bioinformatics/btr131
共享者
- 编辑: 您的姓名。
- 审阅: 审阅者姓名。