# Install packages
if (!requireNamespace("data.table", quietly = TRUE)) {
install.packages("data.table")
}
if (!requireNamespace("jsonlite", quietly = TRUE)) {
install.packages("jsonlite")
}
if (!requireNamespace("ggseqlogo", quietly = TRUE)) {
install.packages("ggseqlogo")
}
if (!requireNamespace("ggplot2", quietly = TRUE)) {
install.packages("ggplot2")
}
# Load packages
library(data.table)
library(jsonlite)
library(ggseqlogo)
library(ggplot2)Seqlogo
Note
Hiplot website
This page is the tutorial for source code version of the Hiplot Seqlogo plugin. You can also use the Hiplot website to achieve no code ploting. For more information please see the following link:
The sequence LOGO is a graphic that describes a sequence pattern of binding sites.
Setup
System Requirements: Cross-platform (Linux/MacOS/Windows)
Programming language: R
Dependent packages:
data.table;jsonlite;ggseqlogo;ggplot2
sessioninfo::session_info("attached")─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.6.0 (2026-04-24)
os Ubuntu 24.04.4 LTS
system x86_64, linux-gnu
ui X11
language (EN)
collate C.UTF-8
ctype C.UTF-8
tz UTC
date 2026-05-09
pandoc 3.1.3 @ /usr/bin/ (via rmarkdown)
quarto 1.9.37 @ /usr/local/bin/quarto
─ Packages ───────────────────────────────────────────────────────────────────
package * version date (UTC) lib source
data.table * 1.18.4 2026-05-06 [1] RSPM
ggplot2 * 4.0.3.9000 2026-05-04 [1] Github (tidyverse/ggplot2@6870419)
ggseqlogo * 0.2.2 2025-12-22 [1] RSPM
jsonlite * 2.0.0 2025-03-27 [1] RSPM
[1] /home/runner/work/_temp/Library
[2] /opt/R/4.6.0/lib/R/site-library
[3] /opt/R/4.6.0/lib/R/library
* ── Packages attached to the search path.
──────────────────────────────────────────────────────────────────────────────Data Preparation
The loaded data is the Sequence of binding sites for multiple transcription factors on multiple genes.
# Load data
data <- data.table::fread(jsonlite::read_json("https://hiplot.cn/ui/basic/ggseqlogo/data.json")$exampleData$textarea[[1]])
data <- as.data.frame(data)
# Convert data structure
data <- data[, !sapply(data, function(x) {all(is.na(x))})]
data <- as.list(data)
data <- lapply(data, function(x) {return(x[!is.na(x)])})
# View data
str(data[1:5])List of 5
$ MA0001.1: chr [1:97] "CCATATATAG" "CCATATATAG" "CCATAAATAG" "CCATAAATAG" ...
$ MA0002.1: chr [1:26] "AATTGTGGTTA" "ATCTGTGGTTA" "AATTGTGGTAA" "TTCTGCGGTTA" ...
$ MA0004.1: chr [1:20] "CACGTG" "CACGTG" "CACGTG" "CACGTG" ...
$ MA0005.1: chr [1:90] "CCTAATTGGGC" "CCTAATTTGGC" "CCTAATCGGGC" "CCTAATCGGGC" ...
$ MA0006.1: chr [1:24] "CGCGTG" "CGCGTG" "CGCGTG" "CGCGTG" ...Visualization
# Seqlogo
p <- ggseqlogo(
data,
ncol = 4,
col_scheme = "nucleotide",
seq_type = "dna",
method = "bits") +
theme(plot.title = element_text(hjust = 0.5))
p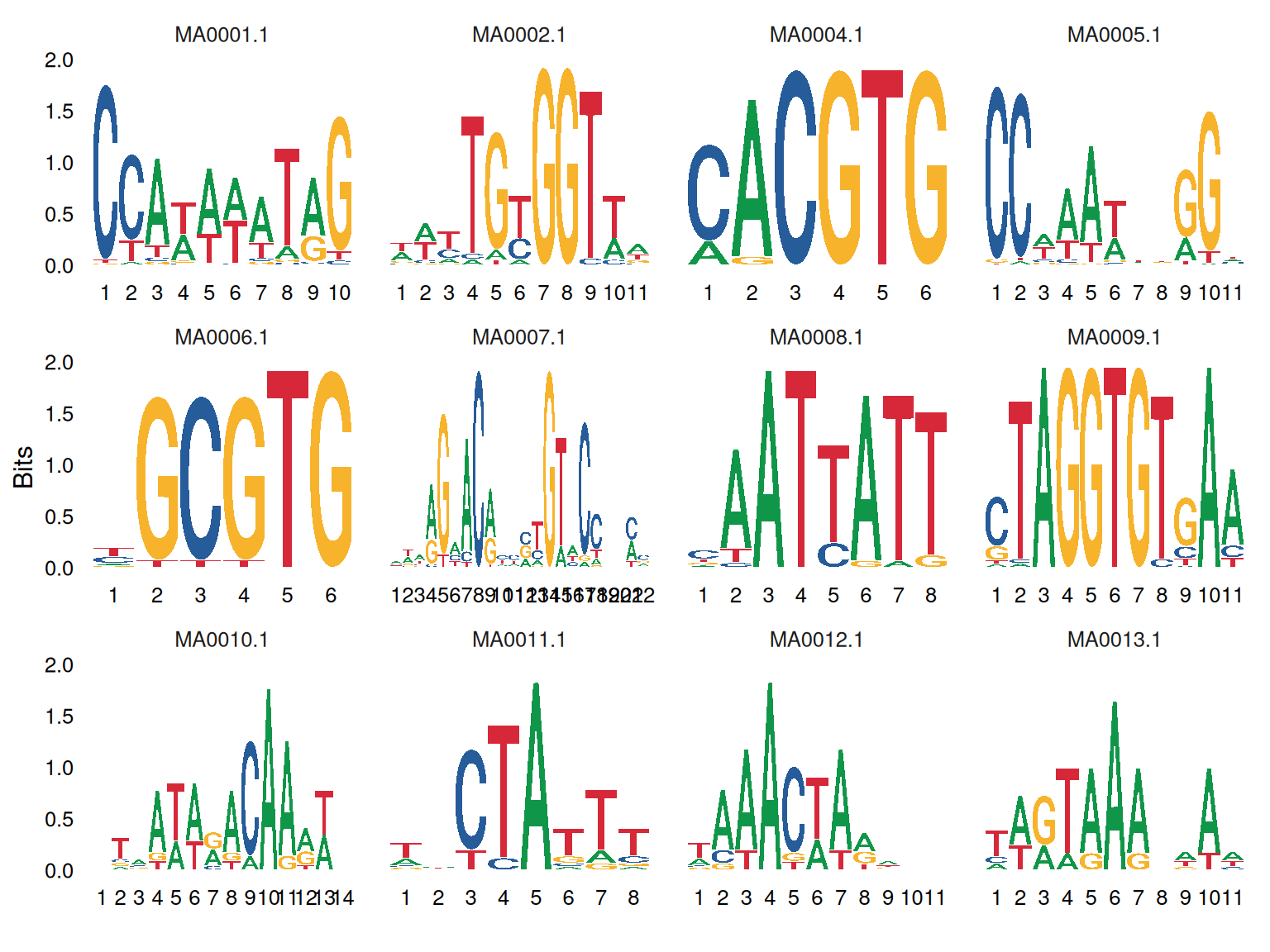
A sequence of binding sites was displayed in a column of the chart by means of BITS calculation, which could clearly observe the large proportion of bases of different sequences.