# Install packages
if (!requireNamespace("data.table", quietly = TRUE)) {
install.packages("data.table")
}
if (!requireNamespace("jsonlite", quietly = TRUE)) {
install.packages("jsonlite")
}
if (!requireNamespace("PCAtools", quietly = TRUE)) {
install.packages('PCAtools', repos = c('https://bioc.r-universe.dev', 'https://cloud.r-project.org'))
}
if (!requireNamespace("ggplotify", quietly = TRUE)) {
install.packages("ggplotify")
}
if (!requireNamespace("cowplot", quietly = TRUE)) {
install.packages("cowplot")
}
# Load packages
library(data.table)
library(jsonlite)
library(PCAtools)
library(ggplotify)
library(cowplot)PCAtools
Note
Hiplot website
This page is the tutorial for source code version of the Hiplot PCAtools plugin. You can also use the Hiplot website to achieve no code ploting. For more information please see the following link:
PCAtools can reduce the dimensionality of data through principal component analysis, and view principal component related features at a two-dimensional level
Setup
System Requirements: Cross-platform (Linux/MacOS/Windows)
Programming language: R
Dependent packages:
data.table;jsonlite;PCAtools;ggplotify;cowplot
sessioninfo::session_info("attached")─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.6.0 (2026-04-24)
os Ubuntu 24.04.4 LTS
system x86_64, linux-gnu
ui X11
language (EN)
collate C.UTF-8
ctype C.UTF-8
tz UTC
date 2026-05-09
pandoc 3.1.3 @ /usr/bin/ (via rmarkdown)
quarto 1.9.37 @ /usr/local/bin/quarto
─ Packages ───────────────────────────────────────────────────────────────────
package * version date (UTC) lib source
cowplot * 1.2.0 2025-07-07 [1] RSPM
data.table * 1.18.4 2026-05-06 [1] RSPM
ggplot2 * 4.0.3.9000 2026-05-04 [1] Github (tidyverse/ggplot2@6870419)
ggplotify * 0.1.3 2025-09-20 [1] RSPM
ggrepel * 0.9.8 2026-03-17 [1] RSPM
jsonlite * 2.0.0 2025-03-27 [1] RSPM
PCAtools * 2.25.0 2026-04-28 [1] https://bioc.r-universe.dev (R 4.6.0)
[1] /home/runner/work/_temp/Library
[2] /opt/R/4.6.0/lib/R/site-library
[3] /opt/R/4.6.0/lib/R/library
* ── Packages attached to the search path.
──────────────────────────────────────────────────────────────────────────────Data Preparation
- Data table 1 (numerical matrix):
Each column is a sample, and each row is a feature (such as gene, chip probe).
- Data sheet 2 (sample information):
The first column is the sample, and the other columns are the phenotypic characteristics of the sample, which can be used to mark the color and shape of the point and perform correlation analysis with the principal component.
# Load data
data <- data.table::fread(jsonlite::read_json("https://hiplot.cn/ui/basic/pcatools/data.json")$exampleData$textarea[[1]])
data <- as.data.frame(data)
data2 <- data.table::fread(jsonlite::read_json("https://hiplot.cn/ui/basic/pcatools/data.json")$exampleData$textarea[[2]])
data2 <- as.data.frame(data2)
# View data
head(data[,1:5]) Probes GSM65752 GSM65753 GSM65755 GSM65757
1 220050_at 6.566843 5.902831 5.185271 5.474453
2 213944_x_at 8.722271 9.088407 9.106401 8.900869
3 215441_at 3.812778 3.852745 3.846690 3.842543
4 214792_x_at 6.499815 6.731196 5.951202 6.578830
5 217251_x_at 6.607354 6.555413 6.715821 6.628053
6 207406_at 3.997302 3.964112 3.836560 3.833057head(data2[,1:5]) Samplename Study Age Distant.RFS ER
1 GSM65752 GSE47561 40 0 ER-
2 GSM65753 GSE47561 46 0 ER+
3 GSM65755 GSE47561 41 1 ER+
4 GSM65757 GSE47561 34 0 ER+
5 GSM65758 GSE47561 46 1 ER+
6 GSM65760 GSE47561 57 1 ER+Visualization
# PCAtools
## Define the plot function
call_pcatools <- function(datTable, sampleInfo,
top_var,
screeplotComponents, screeplotColBar,
pairsplotComponents,
biplotShapeBy, biplotColBy,
plotloadingsComponents,
plotloadingsLowCol,
plotloadingsMidCol,
plotloadingsHighCol,
eigencorplotMetavars,
eigencorplotComponents) {
row.names(datTable) <- datTable[, 1]
datTable <- datTable[, -1]
row.names(sampleInfo) <- sampleInfo[, 1]
data3 <<- pca(datTable, metadata = sampleInfo, removeVar = (100 - top_var) / 100)
for (i in c("screeplotComponents", "pairsplotComponents",
"plotloadingsComponents", "eigencorplotComponents")) {
if (ncol(data3$rotated) < get(i)) {
assign(i, ncol(data3$rotated))
}
}
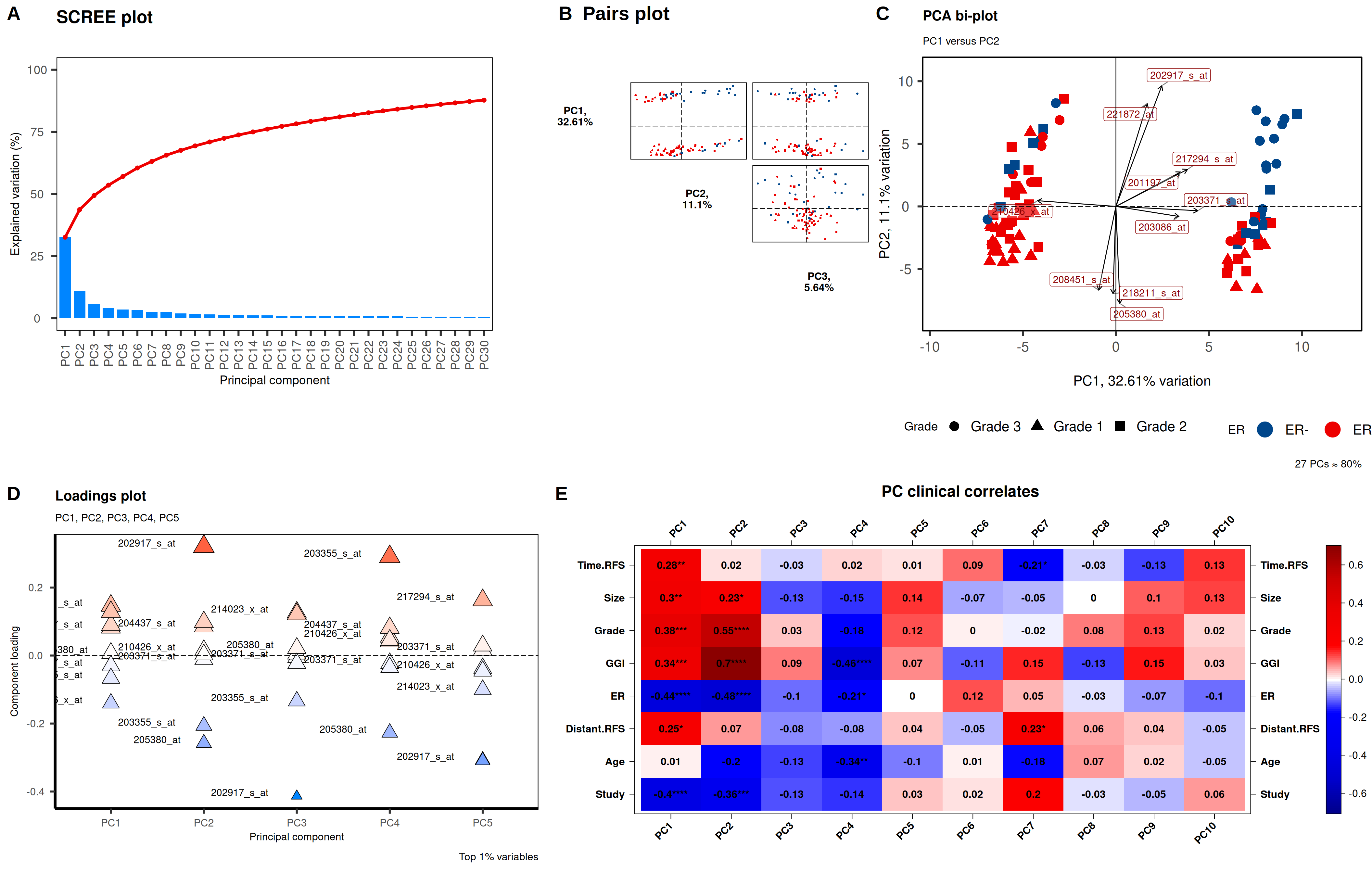
p1 <- PCAtools::screeplot(
data3,
components = getComponents(data3, 1:screeplotComponents),
axisLabSize = 14, titleLabSize = 20,
colBar = screeplotColBar,
gridlines.major = FALSE, gridlines.minor = FALSE,
returnPlot = TRUE
)
params_pairsplot <- list(
data3,
components = getComponents(data3, c(1:pairsplotComponents)),
triangle = TRUE, trianglelabSize = 12,
hline = 0, vline = 0,
pointSize = 0.8, gridlines.major = FALSE, gridlines.minor = FALSE,
title = "", plotaxes = FALSE,
margingaps = unit(c(0.01, 0.01, 0.01, 0.01), "cm"),
returnPlot = TRUE,
colkey = c("#00468BFF","#ED0000FF"),
legendPosition = "none"
)
params_biplot <- list(data3,
showLoadings = TRUE,
lengthLoadingsArrowsFactor = 1.5,
sizeLoadingsNames = 4,
colLoadingsNames = "red4",
# other parameters
lab = NULL,
hline = 0, vline = c(-25, 0, 25),
vlineType = c("dotdash", "solid", "dashed"),
gridlines.major = FALSE, gridlines.minor = FALSE,
pointSize = 5,
legendLabSize = 16, legendIconSize = 8.0,
drawConnectors = FALSE,
title = "PCA bi-plot",
subtitle = "PC1 versus PC2",
caption = "27 PCs ≈ 80%",
returnPlot = TRUE,
legendPosition = "bottom"
)
if (!is.null(biplotShapeBy) && biplotShapeBy != "") {
params_biplot$shape <- biplotShapeBy
params_pairsplot$shape <- biplotShapeBy
t <- params_biplot[[1]]$metadata[,biplotShapeBy]
params_biplot[[1]]$metadata[,biplotShapeBy] <- factor(t,
levels = t[!duplicated(t)]
)
params_pairsplot[[1]]$metadata[,biplotShapeBy] <- factor(t,
levels = t[!duplicated(t)]
)
}
if (!is.null(biplotColBy) && biplotColBy != "") {
params_pairsplot$colby <- biplotColBy
params_pairsplot$colkey <- c("#00468BFF","#ED0000FF")
params_biplot$colby <- biplotColBy
params_biplot$colkey <- c("#00468BFF","#ED0000FF")
t1 <- params_biplot[[1]]$metadata[,biplotColBy]
params_biplot[[1]]$metadata[,biplotColBy] <- factor(t1,
levels = t1[!duplicated(t1)]
)
params_pairsplot[[1]]$metadata[,biplotColBy] <- factor(t1,
levels = t1[!duplicated(t1)]
)
}
p2 <- do.call(PCAtools::pairsplot, params_pairsplot)
p3 <- do.call(PCAtools::biplot, params_biplot)
p4 <- PCAtools::plotloadings(
data3,
rangeRetain = 0.01, labSize = 4,
components = getComponents(data3, c(1:plotloadingsComponents)),
title = "Loadings plot", axisLabSize = 12,
subtitle = "PC1, PC2, PC3, PC4, PC5",
caption = "Top 1% variables",
gridlines.major = FALSE, gridlines.minor = FALSE,
shape = 24, shapeSizeRange = c(4, 8),
col = c(plotloadingsLowCol, plotloadingsMidCol, plotloadingsHighCol),
legendPosition = "none",
drawConnectors = FALSE,
returnPlot = TRUE
)
if (length(eigencorplotMetavars) > 0 && all(eigencorplotMetavars != "")) {
metavars <- eigencorplotMetavars
} else {
metavars <- colnames(sampleInfo)[2:ncol(sampleInfo)]
}
if (length(metavars) == 1 && metavars != colnames(sampleInfo)[1]) {
metavars <- c(colnames(sampleInfo)[1], metavars)
} else if (length(metavars) == 1 && metavars == colnames(sampleInfo)[1]) {
stop('eigencorplotMetavars need >= 2 feature')
}
p5 <- PCAtools::eigencorplot(
data3,
components = getComponents(data3, 1:eigencorplotComponents),
metavars = metavars,
cexCorval = 1.0,
fontCorval = 2,
posLab = "all",
rotLabX = 45,
scale = TRUE,
main = "PC clinical correlates",
cexMain = 1.5,
plotRsquared = FALSE,
corFUN = "pearson",
corUSE = "na.or.complete",
signifSymbols = c("****", "***", "**", "*", ""),
signifCutpoints = c(0, 0.0001, 0.001, 0.01, 0.05, 1),
returnPlot = TRUE
)
p6 <- plot_grid(
p1, p2, p3,
ncol = 3,
labels = c("A", "B Pairs plot", "C"),
label_fontfamily = "Arial",
label_fontface = "bold",
label_size = 22,
align = "h",
rel_widths = c(1.10, 0.80, 1.10)
)
p7 <- plot_grid(
p4,
as.grob(p5),
ncol = 2,
labels = c("D", "E"),
label_fontfamily = "Arial",
label_fontface = "bold",
label_size = 22,
align = "h",
rel_widths = c(0.8, 1.2)
)
p <- plot_grid(
p6, p7,
ncol = 1,
rel_heights = c(1.1, 0.9)
)
return(p)
}
## plot
p <- call_pcatools(
datTable = data,
sampleInfo = data2,
biplotColBy = "ER",
biplotShapeBy = "Grade",
eigencorplotMetavars = colnames(data2)[-1],
screeplotComponents = 30,
pairsplotComponents = 3,
plotloadingsComponents = 5,
eigencorplotComponents = 10,
top_var = 90,
screeplotColBar = "#0085FF",
plotloadingsLowCol = "#0085FF",
plotloadingsMidCol = "#FFFFFF",
plotloadingsHighCol = "#FF0000"
)
p