# 安装包
if (!requireNamespace("tidyverse", quietly = TRUE)){
install.packages("tidyverse")
}
if (!requireNamespace("ggprism", quietly = TRUE)){
install.packages("ggprism")
}
if (!requireNamespace("gground", quietly = TRUE)) {
remotes::install_github("dxsbiocc/gground")
}
# 加载包
library(tidyverse)
library(ggprism)
library(gground)文字叠加的富集分析柱状图
文字叠加富集柱状图是一种用于高密度展示功能富集分析结果(如 GO、KEGG)的可视化工具。它通常以圆角柱状图的长度映射富集显著性(adjust p value),并利用图形内部空间直接叠加标注通路名称与核心基因列表,同时辅以左侧色块与气泡来区分功能分类及基因数量。
这种图表形态改变了传统条形图文本与图形分离的布局,特别适用于单细胞亚群标记基因的功能注释展示,以及其他需要同时呈现“通路显著性”与“具体驱动基因”的研究场景。其可视化效果不仅优化了绘图空间利用率,还便于研究者在概览生物学功能的同时,快速锁定导致通路富集的关键基因特征。
Example

环境配置
系统要求 跨平台(Linux/MacOS/Windows)
编程语言: R
依赖包:
tidyverse,ggprism,gground
数据准备
1.上游富集分析流程 (参考)
在实际的生物信息学分析中,我们通常使用 clusterProfiler 进行富集分析。
注记
以下代码仅用于演示上游分析流程(获取原始数据),本教程绘图将直接使用下文的示例数据。
# 1. 环境准备
library(clusterProfiler)
library(org.Hs.eg.db)
#2. 运行富集分析
# 假设 gene_list 是差异基因的 Entrez ID 向量
# 2.1 GO 富集分析 (同时获取 BP/CC/MF)
ego <- enrichGO(
gene = gene_list,
OrgDb = org.Hs.eg.db,
ont = "ALL",
readable = TRUE # 自动将结果中的 ID 转换为 Symbol
)
# 2.2 KEGG 富集分析
ekegg <- enrichKEGG(
gene = gene_list,
organism = 'hsa'
)
# KEGG 结果默认为 Entrez ID,需手动转为 Symbol 以匹配绘图数据格式
ekegg <- setReadable(ekegg, OrgDb = org.Hs.eg.db, keyType = "ENTREZID")
# 3. 数据清洗与合并
# 提取 GO 结果 (重命名 ONTOLOGY 为 Category)
df_go <- as.data.frame(ego) %>%
select(Category = ONTOLOGY, Description, p.adjust, Count, geneID)
# 提取 KEGG 结果 (手动添加 Category 列并赋值为 "KEGG")
df_kegg <- as.data.frame(ekegg) %>%
mutate(Category = "KEGG") %>%
select(Category, Description, p.adjust, Count, geneID)
# 合并所有结果 -> 得到最终用于绘图的 raw_data
raw_data <- rbind(df_go, df_kegg)2.示例数据加载与清洗
为了方便教程运行,我们导入来自 DAVID 的网络药理学注释分析通路富集数据,该数据包含了 GO (BP/CC/MF) 和 KEGG 的富集结果。
#1.读取数据
raw_data <- read_tsv("https://bizard-1301043367.cos.ap-guangzhou.myqcloud.com/DAVID.txt")
# 2. 数据清洗
# 将 DAVID 的格式转换为绘图所需的标准格式
raw_data <- raw_data %>%
# 2.1 提取分类标签
mutate(Category = case_when(
grepl("BP_DIRECT", Category) ~ "BP",
grepl("CC_DIRECT", Category) ~ "CC",
grepl("MF_DIRECT", Category) ~ "MF",
grepl("KEGG_PATHWAY", Category) ~ "KEGG",
TRUE ~ "Other"
)) %>%
# 只保留GO、KEGG结果
filter(Category %in% c("BP", "CC", "MF", "KEGG")) %>%
# 2.2 清洗通路名称 (去掉 ID)
mutate(Description = sub("^.*~|.*:", "", Term)) %>%
# 2.3 重命名列
rename(
p.adjust = FDR,
geneID = Genes
) %>%
# 2.4 格式化基因列表
mutate(geneID = gsub(", ", "/", geneID)) 3.创建绘图数据
# 1.筛选每个分类下最显著的 Top 5 通路
use_pathway <- raw_data %>%
arrange(p.adjust) %>%
group_by(Category) %>%
slice_head(n = 5) %>%
ungroup()
# 2. 构造绘图辅助列
use_pathway <- use_pathway %>%
# 设置因子水平:决定图例和绘图的垂直顺序
mutate(Category = factor(Category, levels = rev(c('BP', 'CC', 'MF', 'KEGG')))) %>%
# 二次排序
arrange(Category, p.adjust) %>%
# 固定 Y 轴顺序
mutate(Description = factor(Description, levels = Description)) %>%
# 添加 index 列作为 Y 轴坐标
tibble::rowid_to_column('index')
# 3. 构造左侧分类色块的数据 (rect.data)
#布局参数
width <- 0.5
# 计算 X 轴最大长度,用于绘制底部横线
xaxis_max <- max(-log10(use_pathway$p.adjust)) + 1
rect.data <- use_pathway %>%
arrange(Category) %>%
group_by(Category) %>%
summarize(n = n(), .groups = "drop") %>%
mutate(
xmin = -3 * width,
xmax = -2 * width,
ymax = cumsum(n),
ymin = lag(ymax, default = 0) + 0.6,
ymax = ymax + 0.4
)
# 查看数据结构
head(use_pathway)# A tibble: 6 × 15
index Category Term Count `%` PValue geneID `List Total` `Pop Hits`
<int> <fct> <chr> <dbl> <dbl> <dbl> <chr> <dbl> <dbl>
1 1 KEGG hsa04080:N… 15 36.6 2.07e-10 GABRB… 40 368
2 2 KEGG hsa04726:S… 10 24.4 1.16e- 9 GABRB… 40 115
3 3 KEGG hsa05022:P… 11 26.8 3.19e- 5 IL1A/… 40 483
4 4 KEGG hsa05321:I… 5 12.2 1.89e- 4 IL10/… 40 66
5 5 KEGG hsa04728:D… 6 14.6 2.6 e- 4 GSK3B… 40 132
6 6 MF GO:0051378… 6 14.6 2.37e-11 HTR1A… 41 12
# ℹ 6 more variables: `Pop Total` <dbl>, FoldEnrichment <dbl>,
# Bonferroni <dbl>, Benjamini <dbl>, p.adjust <dbl>, Description <fct>可视化
1. 基础绘图(简化版)
该版本主要展示了核心的圆角柱状图和左侧的分类色块,去除了基因列表等过多文本信息,适合需要简洁展示的场景。
# 定义配色
pal <- c('#eaa052', '#b74147', '#90ad5b', '#23929c')
#其它推荐配色
#pal <- c('#c3e1e6', '#f3dfb7', '#dcc6dc', '#96c38e')
#pal <- c('#7bc4e2', '#acd372', '#fbb05b', '#ed6ca4')
# 调整简化版的左侧色块位置参数
rect.data.simple <- rect.data %>%
mutate(
xmin = -1.5 * width,
xmax = -0.5 * width
)
p1 <- ggplot(use_pathway, aes(-log10(p.adjust), y = index, fill = Category)) +
# 1. 圆角柱状图主体
geom_round_col(aes(y = Description), width = 0.6, alpha = 0.8) +
# 2. 通路名称文本
geom_text(aes(x = 0.05, label = Description), hjust = 0, size = 4) +
# 3. 左侧分类色块
geom_round_rect(
aes(xmin = xmin, xmax = xmax, ymin = ymin, ymax = ymax, fill = Category),
data = rect.data.simple,
radius = unit(2, 'mm'),
inherit.aes = FALSE
) +
# 4. 左侧分类文字
geom_text(
aes(x = (xmin + xmax) / 2, y = (ymin + ymax) / 2, label = Category),
data = rect.data.simple,
size = 3,
inherit.aes = FALSE
) +
# 5. 底部装饰横线
geom_segment(
aes(x = 0, y = 0, xend = xaxis_max, yend = 0),
linewidth = 1,
inherit.aes = FALSE
) +
# 6. 样式调整
labs(y = NULL, x = "-log10(p.adjust)") +
scale_fill_manual(name = 'Category', values = pal) +
scale_x_continuous(breaks = seq(0, xaxis_max, 2), expand = expansion(c(0, 0.1))) +
theme_prism() +
theme(
axis.text.y = element_blank(),
axis.line.y = element_blank(),
axis.ticks.y = element_blank(),
axis.line.x = element_blank(), # 隐藏系统自带的 X 轴线,只保留我们用 geom_segment 画的那条
legend.title = element_text(),
legend.position = "right"
)
p1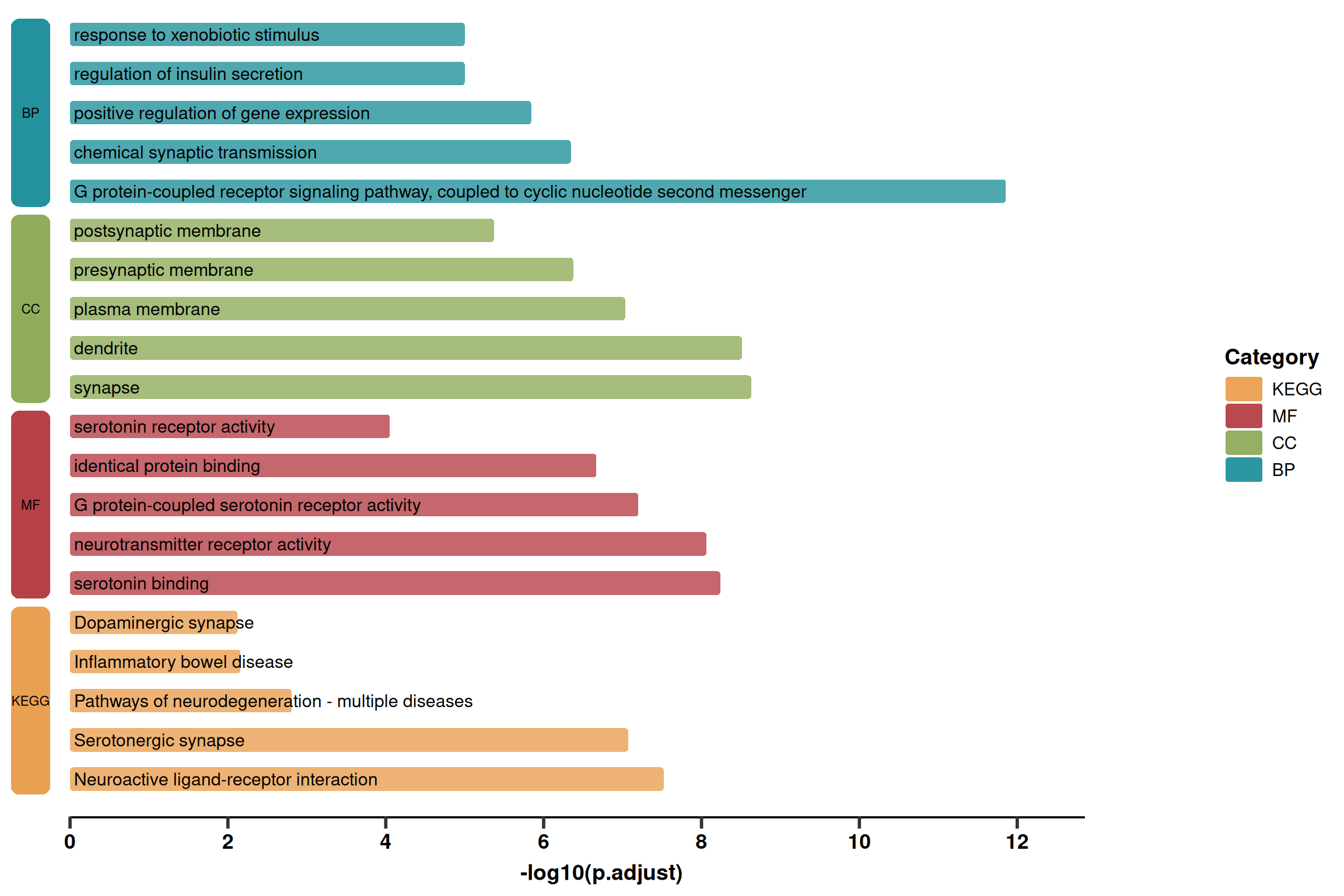
2. 进阶绘图 (详细信息版)
该版本在简化版的基础上,增加了:
Gene List: 在通路名称下方显示基因列表(灰色斜体)。
Count Bubbles: 在左侧展示富集基因的数量(气泡大小)。
Layout: 调整了左侧色块的位置,为气泡留出空间。
p2 <- ggplot(use_pathway, aes(-log10(p.adjust), y = index, fill = Category)) +
# 1. 主体圆角柱
geom_round_col(aes(y = Description), width = 0.6, alpha = 0.8) +
# 2. 通路名称
geom_text(aes(x = 0.05, label = Description), hjust = 0, size = 5) +
# 3. 基因列表
geom_text(
aes(x = 0.1, label = geneID, colour = Category),
hjust = 0, vjust = 2.6, size = 3, fontface = 'italic',
show.legend = FALSE
) +
# 4. 左侧基因计数气泡
geom_point(aes(x = -width, size = Count,fill = Category), shape = 21, stroke = 1) +
geom_text(aes(x = -width, label = Count), size = 3) +
# 5. 左侧分类色块
geom_round_rect(
aes(xmin = xmin, xmax = xmax, ymin = ymin, ymax = ymax, fill = Category),
data = rect.data,
radius = unit(2, 'mm'),
inherit.aes = FALSE
) +
geom_text(
aes(x = (xmin + xmax) / 2, y = (ymin + ymax) / 2, label = Category),
data = rect.data,
inherit.aes = FALSE,
color = "black", fontface = "bold"
) +
# 6. 底部横线
geom_segment(
aes(x = 0, y = 0, xend = xaxis_max, yend = 0),
linewidth = 1.5,
inherit.aes = FALSE
) +
# 7. 标尺与配色
labs(y = NULL, x = "-log10(p.adjust)") +
scale_fill_manual(name = 'Category', values = pal) +
scale_colour_manual(values = pal) +
scale_size_continuous(name = 'Count', range = c(4, 10)) +
scale_x_continuous(breaks = seq(0, xaxis_max, 2), expand = expansion(c(0, 0))) +
# 8. 主题美化
theme_prism() +
theme(
axis.text.y = element_blank(),
axis.line = element_blank(),
axis.ticks.y = element_blank(),
legend.title = element_text(),
plot.margin = margin(l = 10, r = 10)
)
p2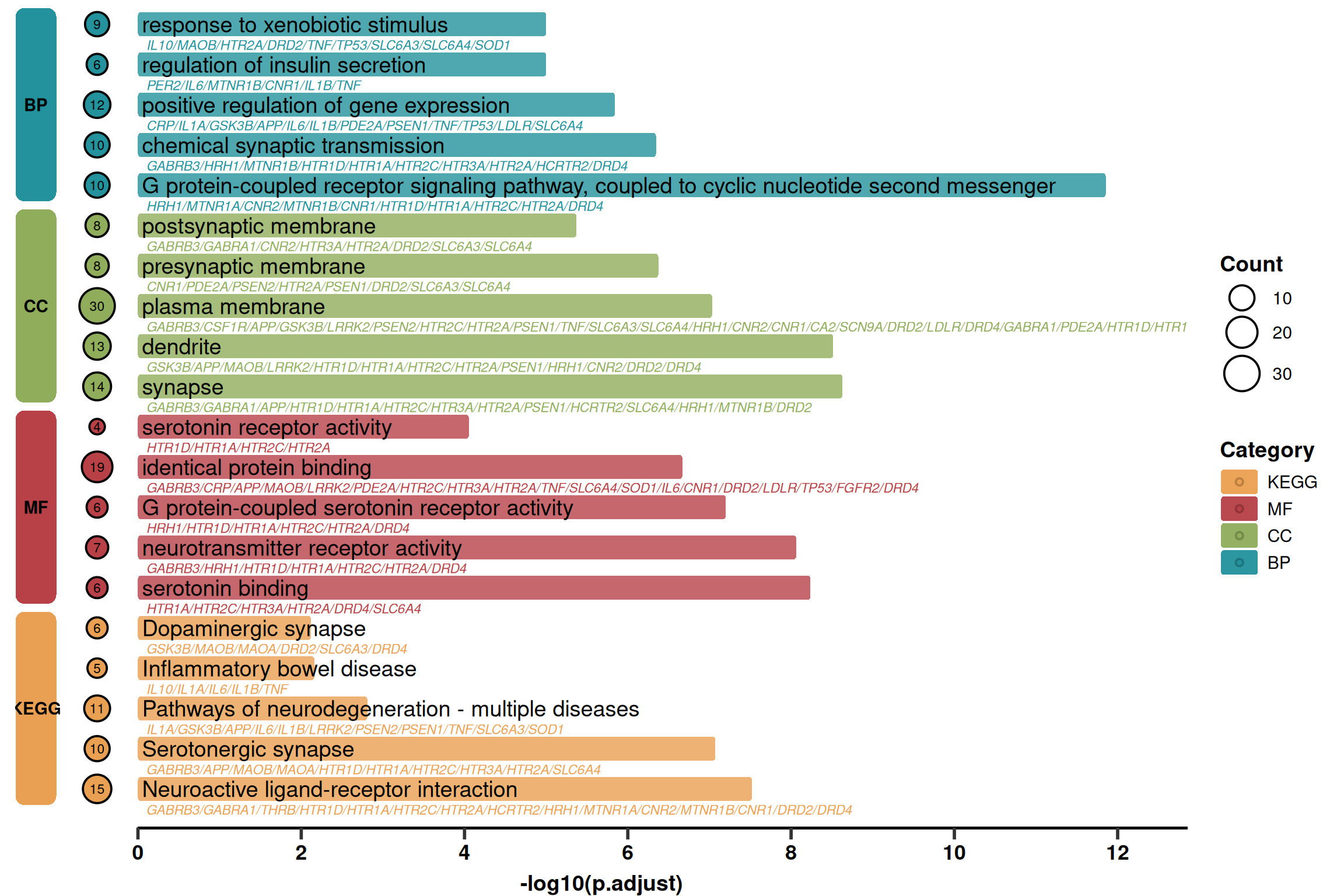
参考资料
[1] 生信医道. 超好看的单细胞GO,KEGG富集分析图!确定不来看看吗? [Internet]. 微信公众号: 生信医道. 2025 [cited 2025 Dec 30]. Available from: https://mp.weixin.qq.com/s/pY1S0EJQAhdezvxeNt-SzA