# 安装包
if (!requireNamespace("data.table", quietly = TRUE)) {
install.packages("data.table")
}
if (!requireNamespace("jsonlite", quietly = TRUE)) {
install.packages("jsonlite")
}
if (!requireNamespace("umap", quietly = TRUE)) {
install.packages("umap")
}
if (!requireNamespace("ggpubr", quietly = TRUE)) {
install.packages("ggpubr")
}
# 加载包
library(data.table)
library(jsonlite)
library(umap)
library(ggpubr)UMAP
注记
Hiplot 网站
本页面为 Hiplot UMAP 插件的源码版本教程,您也可以使用 Hiplot 网站实现无代码绘图,更多信息请查看以下链接:
UMAP 是一种非线性降维算法,适用于高维数据降维到 2 维或 3 维并进行可视化。该算法能够使较大相似度的点,t 分布在低维空间中的距离更近;而对于低相似度的点,t 分布在低维空间中的距离更远。
环境配置
系统: Cross-platform (Linux/MacOS/Windows)
编程语言: R
依赖包:
data.table;jsonlite;umap;ggpubr
sessioninfo::session_info("attached")─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.6.0 (2026-04-24)
os Ubuntu 24.04.4 LTS
system x86_64, linux-gnu
ui X11
language (EN)
collate C.UTF-8
ctype C.UTF-8
tz UTC
date 2026-05-09
pandoc 3.1.3 @ /usr/bin/ (via rmarkdown)
quarto 1.9.37 @ /usr/local/bin/quarto
─ Packages ───────────────────────────────────────────────────────────────────
package * version date (UTC) lib source
data.table * 1.18.4 2026-05-06 [1] RSPM
ggplot2 * 4.0.3.9000 2026-05-04 [1] Github (tidyverse/ggplot2@6870419)
ggpubr * 0.6.3 2026-02-24 [1] RSPM
jsonlite * 2.0.0 2025-03-27 [1] RSPM
umap * 0.2.10.0 2023-02-01 [1] RSPM
[1] /home/runner/work/_temp/Library
[2] /opt/R/4.6.0/lib/R/site-library
[3] /opt/R/4.6.0/lib/R/library
* ── Packages attached to the search path.
──────────────────────────────────────────────────────────────────────────────数据准备
载入数据为数据集(基因名称及其对应的基因表达值)和样本信息(样本名称及分组)。
# 加载数据
data1 <- data.table::fread(jsonlite::read_json("https://hiplot.cn/ui/basic/umap/data.json")$exampleData$textarea[[1]])
data1 <- as.data.frame(data1)
data2 <- data.table::fread(jsonlite::read_json("https://hiplot.cn/ui/basic/umap/data.json")$exampleData$textarea[[2]])
data2 <- as.data.frame(data2)
# 整理数据格式
sample.info <- data2
rownames(data1) <- data1[, 1]
data1 <- as.matrix(data1[, -1])
## umap
set.seed(123)
umap_info <- umap(t(data1))
colnames(umap_info$layout) <- c("UMAP_1", "UMAP_2")
# handle data
umap_data <- data.frame(
sample = colnames(data1),
umap_info$layout
)
colorBy <- sample.info[match(colnames(data1), sample.info[, 1]), "Species"]
colorBy <- factor(colorBy, level = colorBy[!duplicated(colorBy)])
umap_data$colorBy = colorBy
shapeBy <- NULL
# 查看数据
head(data1[,1:5]) M1 M2 M3 M4 M5
Sepal.Length 5.1 4.9 4.7 4.6 5.0
Sepal.Width 3.5 3.0 3.2 3.1 3.6
Petal.Length 1.4 1.4 1.3 1.5 1.4
Petal.Width 0.2 0.2 0.2 0.2 0.2head(data2) Samples Species
1 M1 setosa
2 M2 setosa
3 M3 setosa
4 M4 setosa
5 M5 setosa
6 M6 setosa可视化
# umap
p <- ggscatter(data = umap_data, x = "UMAP_1", y = "UMAP_2", size = 2,
palette = "lancet", color = "colorBy") +
labs(color = "group") +
ggtitle("UMAP Plot") +
theme_classic() +
theme(text = element_text(family = "Arial"),
plot.title = element_text(size = 12,hjust = 0.5),
axis.title = element_text(size = 12),
axis.text = element_text(size = 10),
axis.text.x = element_text(angle = 0, hjust = 0.5,vjust = 1),
legend.position = "right",
legend.direction = "vertical",
legend.title = element_text(size = 10),
legend.text = element_text(size = 10))
p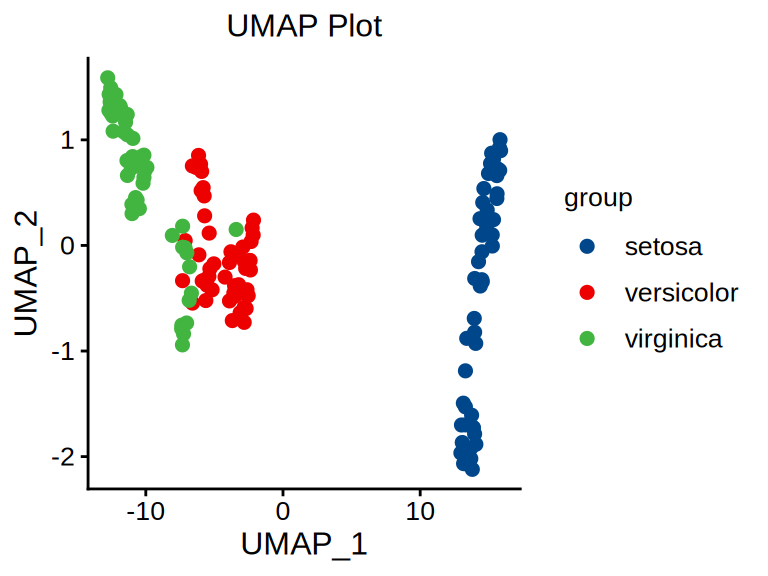
不同颜色表示不同样本,与 PCA(主成分分析)图形解释相同,不同之处在于可视化效果,t-SNE 中对于不相似的点,用一个较小的距离会产生较大的梯度来让这些点排斥开来。