# 安装包
if (!requireNamespace("ggplot2", quietly = TRUE)) {
install.packages("ggplot2")
}
if (!requireNamespace("ggseqlogo", quietly = TRUE)) {
install.packages("ggseqlogo")
}
if (!requireNamespace("cowplot", quietly = TRUE)) {
install.packages("cowplot")
}
if (!requireNamespace("gridExtra", quietly = TRUE)) {
install.packages("gridExtra")
}
# 加载包
library(ggplot2)
library(ggseqlogo)
library(cowplot)
library(gridExtra)Motif图
对于motif logo图的可视化,ggseqlogo是一个基于ggplot2的R包,专门用于绘制序列motif的logo图。相比于其他motif可视化工具,ggseqlogo具有语法简洁、输出格式灵活、与ggplot2生态系统完全兼容等优点。该包支持多种序列格式输入,包括位置频率矩阵(PFM)、位置权重矩阵(PWM)和序列向量,并提供了丰富的自定义选项来调整logo图的外观。
示例

Motif logo图是用于展示DNA、RNA或蛋白质序列中保守模式的图形,通过每个位置的字符大小来显示该位置的信息含量。
环境配置
系统要求: 跨平台(Linux/MacOS/Windows)
编程语言:R
依赖包:
ggplot2,ggseqlogo,cowplot,gridExtra
sessioninfo::session_info("attached")─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.6.0 (2026-04-24)
os Ubuntu 24.04.4 LTS
system x86_64, linux-gnu
ui X11
language (EN)
collate C.UTF-8
ctype C.UTF-8
tz UTC
date 2026-05-09
pandoc 3.1.3 @ /usr/bin/ (via rmarkdown)
quarto 1.9.37 @ /usr/local/bin/quarto
─ Packages ───────────────────────────────────────────────────────────────────
package * version date (UTC) lib source
cowplot * 1.2.0 2025-07-07 [1] RSPM
ggplot2 * 4.0.3.9000 2026-05-04 [1] Github (tidyverse/ggplot2@6870419)
ggseqlogo * 0.2.2 2025-12-22 [1] RSPM
gridExtra * 2.3 2017-09-09 [1] RSPM
[1] /home/runner/work/_temp/Library
[2] /opt/R/4.6.0/lib/R/site-library
[3] /opt/R/4.6.0/lib/R/library
* ── Packages attached to the search path.
──────────────────────────────────────────────────────────────────────────────数据准备
使用内置数据集 ggseqlogo_sample,该数据集包含三个变量、两种不同的输入形式: - seqs_dna:从JASPAR的FASTA文件中获得的12个转录因子的结合位点。格式为字符串向量组成的list,list的名称表示JASPAR ID。 - seqs_aa:从Wagih等人获得的激酶底物磷酸化位点组。格式与seqs_dna相同。 - pfms_dna:从JASPAR获得的四个转录因子的位置频率matrix组成的list,list的名称表示JASPAR ID。
data(ggseqlogo_sample)
head(pfms_dna,n = 1)$MA0018.2
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8]
A 0 0 11 0 1 0 2 8
C 1 1 0 9 0 3 7 0
G 1 10 0 2 10 0 1 1
T 9 0 0 0 0 8 1 2head(seqs_aa, n = 1)[[1]][1:3][1] "VVGARRSSWRVVSSI" "GPRSRSRSRDRRRKE" "LLCLRRSSLKAYGNG"可视化
1. 基础Motif图
ggseqlogo 包可以使用 geom_logo 函数并基于 ggplot2 语法进行绘制,也可以使用封装后的函数 ggseqlogo 函数进行绘制,二者共享相同的参数。
# 使用序列向量
ggseqlogo(seqs_dna$MA0001.1)
# 使用PFM矩阵
ggseqlogo(pfms_dna$MA0018.2)
#使用ggplot语法绘图
ggplot() + geom_logo( seqs_dna$MA0001.1 ) + theme_logo()


上图为MA0001.1的motif结果展示
提示
关键参数:
- data:输入数据:序列向量、矩阵或列表
- method = “bits”:计算方法:“bits”或”probability”
- seq_type = “auto”:序列类型:“auto”、“dna”、“rna”、“aa”
- namespace = NULL:自定义字符命名空间
- font = “roboto_medium”:字体类型
- stack_width = 0.95:字符堆叠宽度
- rev_stack_order = FALSE:是否反转堆叠顺序
- col_scheme = “auto”:颜色方案
- low_col = ‘black’:低bits/probability颜色
- high_col = ‘yellow’:高bits/probability颜色
- na_col = ‘grey20’:NA值颜色
- plot = TRUE:是否立即绘图
2. 多motif绘图
你可以使用facet_wrap或facet_grid来组合多个logo图:
# 绘制多个motif
ggseqlogo(seqs_dna, ncol=4)
# 等价于
p <- ggplot() + geom_logo(seqs_dna) + theme_logo() +
facet_wrap(~seq_group, ncol=4, scales='free_x')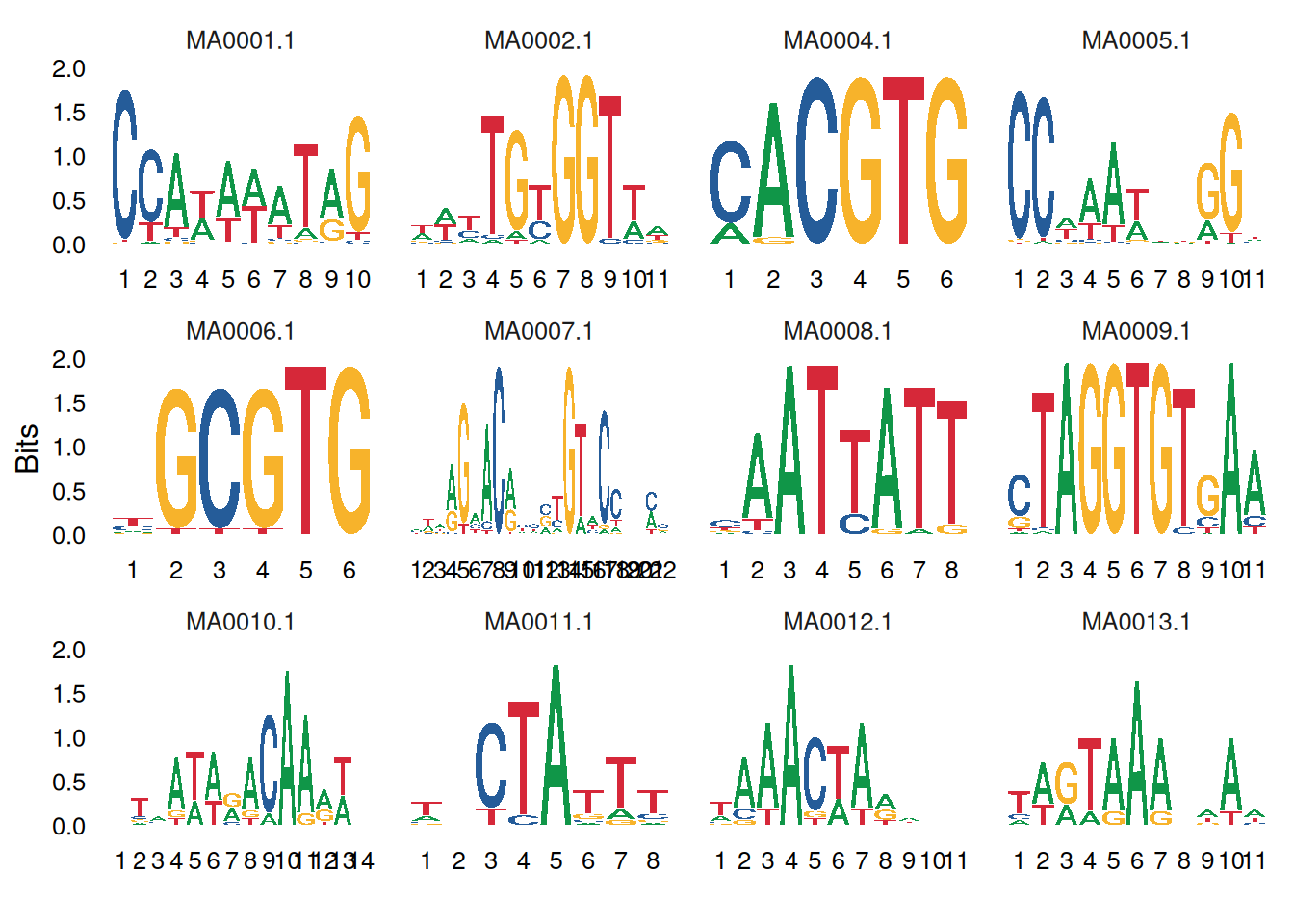
3. Motif图美化
3.1 调整颜色方案
ggseqlogo 提供了多种预设和自定义颜色方案:
# DNA序列的预设颜色方案
ggseqlogo(seqs_dna$MA0001.1, col_scheme='nucleotide')
# 氨基酸序列的颜色方案
ggseqlogo(seqs_aa$AKT1, col_scheme='chemistry')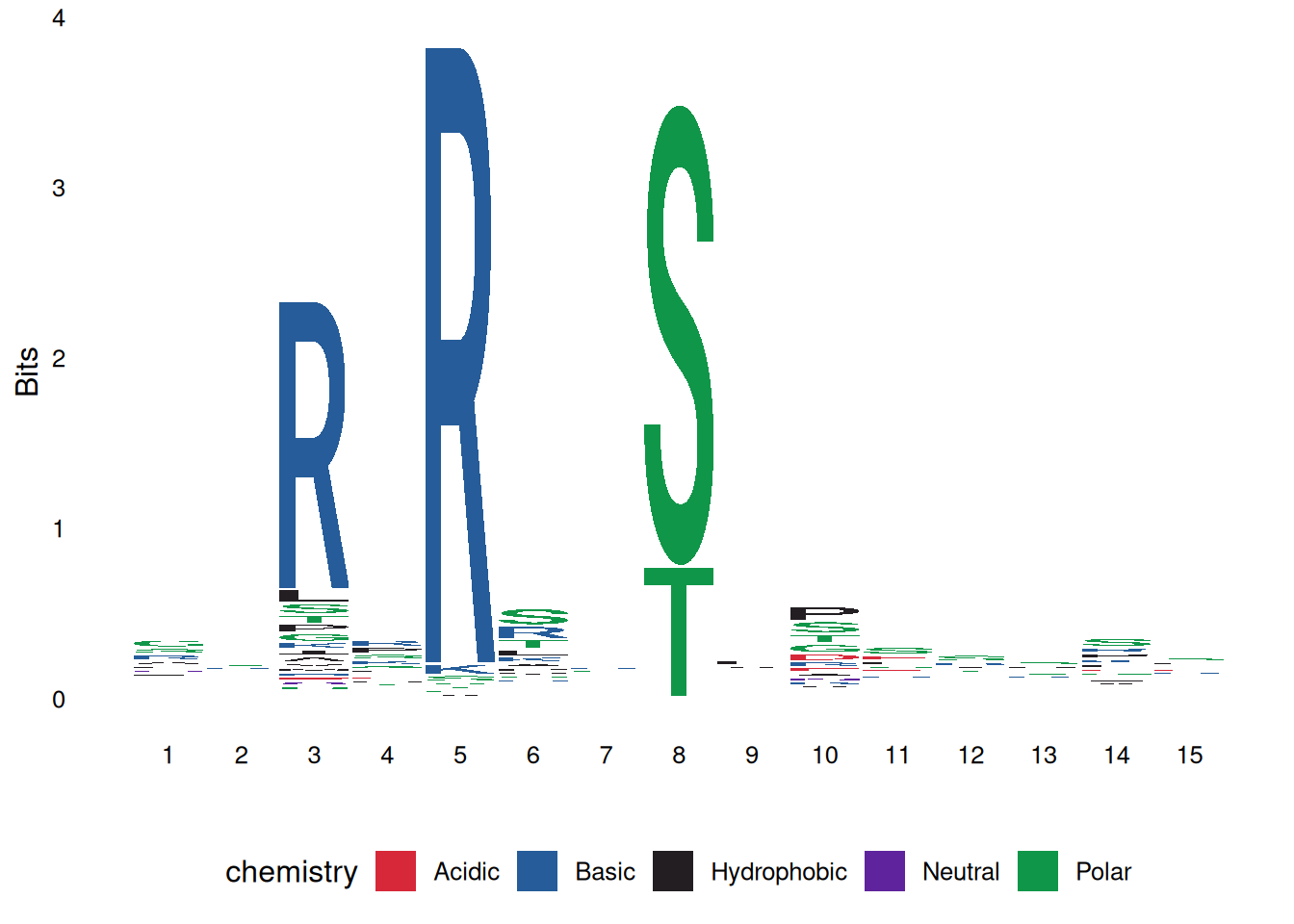
# 自定义离散颜色方案
cs1 <- make_col_scheme(chars=c('A', 'T', 'C', 'G'),
groups=c('gr1', 'gr1', 'gr2', 'gr2'),
cols=c('purple', 'purple', 'blue', 'blue'))
ggseqlogo(seqs_dna$MA0001.1, col_scheme=cs1)
# 自定义连续颜色方案
cs2 <- make_col_scheme(chars=c('A', 'T', 'C', 'G'), values=1:4)
ggseqlogo(seqs_dna$MA0001.1, col_scheme=cs2)
3.2 调整字体和堆叠
# 查看所有可用字体
list_fonts(F) [1] "helvetica_regular" "helvetica_bold" "helvetica_light"
[4] "roboto_medium" "roboto_bold" "roboto_regular"
[7] "akrobat_bold" "akrobat_regular" "roboto_slab_bold"
[10] "roboto_slab_regular" "roboto_slab_light" "xkcd_regular" # 使用特定字体
ggseqlogo(seqs_dna$MA0001.1, font='helvetica_bold', stack_width=0.8)
3.3 调整坐标轴和主题
ggseqlogo(seqs_dna$MA0001.1) +
theme_classic() +
theme(axis.text.x = element_text(angle=45, hjust=1)) +
labs(x='Position', y='Bits', title='Transcription Factor Binding Motif')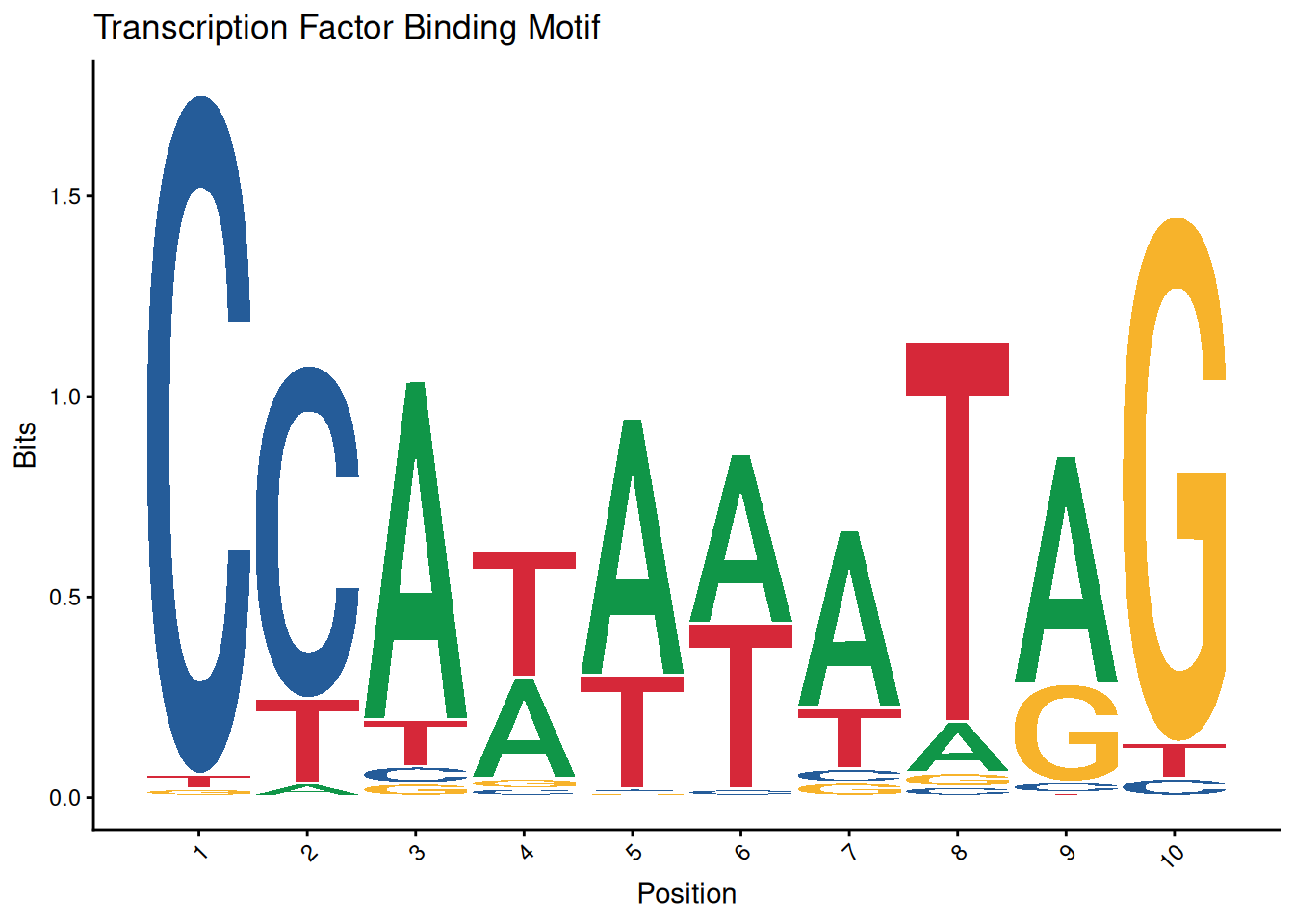
4. 高级功能
绘图方法选择:
ggseqlogo 支持两种序列 logo 计算方法:
p1 <- ggseqlogo(seqs_dna$MA0001.1, method='bits') # 信息量
p2 <- ggseqlogo(seqs_dna$MA0001.1, method='prob') # 概率
gridExtra::grid.arrange(p1, p2, ncol=2)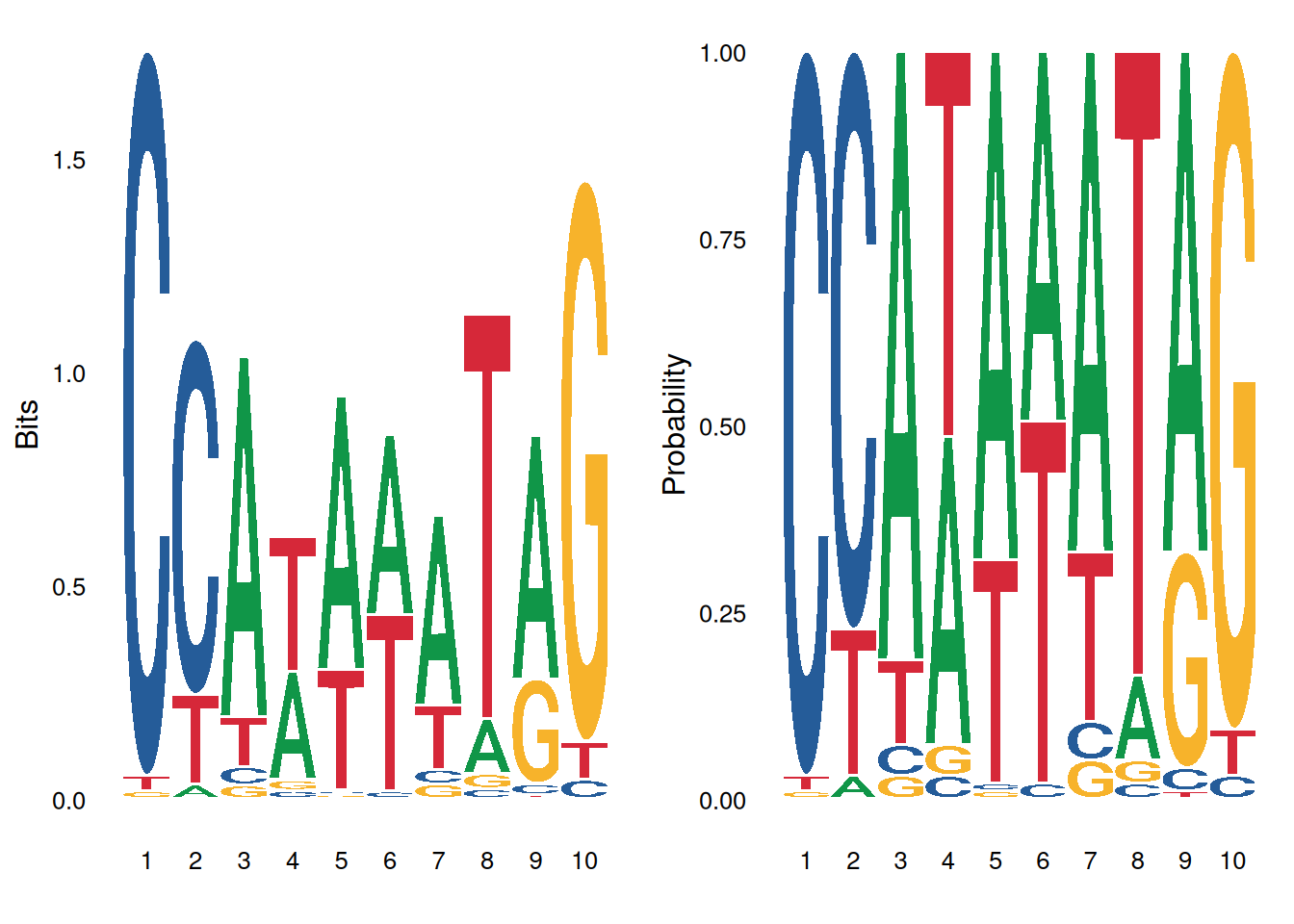
自定义序列类型和命名空间:
# 数值序列
seqs_numeric <- chartr('ATGC', '1234', seqs_dna$MA0001.1)
ggseqlogo(seqs_numeric, method='prob', namespace=1:4)
# 希腊字母序列
seqs_greek <- chartr('ATGC', 'δεψλ', seqs_dna$MA0001.1)
ggseqlogo(seqs_greek, namespace='δεψλ', method='bits')
自定义高度logo:
# 创建自定义高度矩阵
custom_mat <- matrix(rnorm(20), nrow=4,
dimnames=list(c('A', 'T', 'G', 'C')))
ggseqlogo(custom_mat, method='custom', seq_type='dna') +
ylab('my custom height')
序列标识:
ggplot() +
annotate('rect', xmin=0.5, xmax=3.5, ymin=-0.05, ymax=1.9,
alpha=0.1, col='black', fill='yellow') +
geom_logo(seqs_dna$MA0001.1, stack_width=0.90) +
annotate('segment', x=4, xend=8, y=1.2, yend=1.2, size=2) + #需要注意的是在ggplot2 3.4.0版本开始,调整线条粗细的size参数已经改变为了linewidth参数,建议使用新版本ggplot2的用户将size改为linewidth。
annotate('text', x=6, y=1.3, label='Text annotation') +
theme_logo()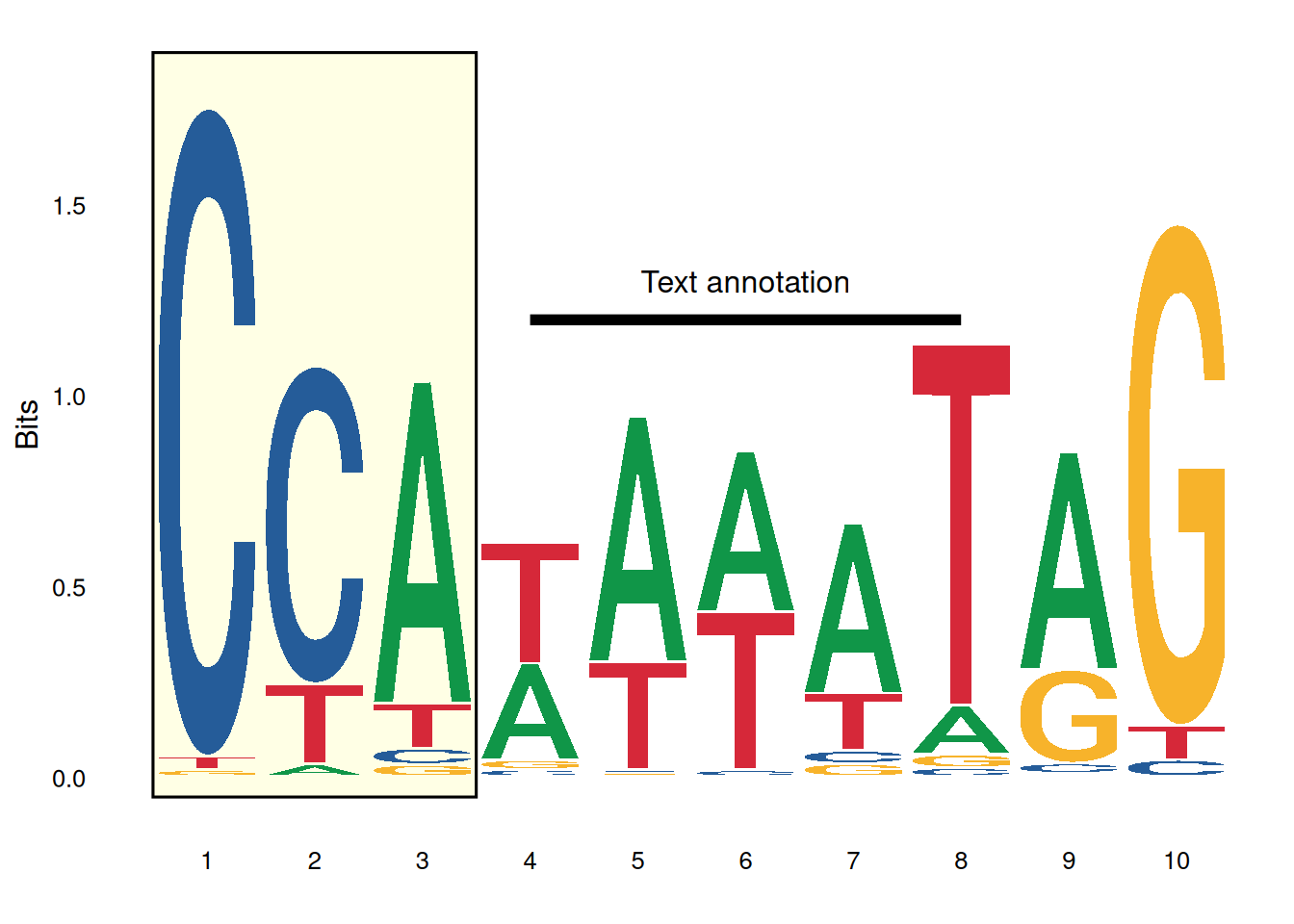
组合多种图形:
# 生成序列logo
p1 <- ggseqlogo(seqs_dna$MA0008.1) +
theme(axis.text.x=element_blank())
# 创建序列比对数据
aln <- data.frame(
letter=strsplit('AGATAAGATGATAAAAAGATAAGA', '')[[1]],
species=rep(c('a', 'b', 'c'), each=8),
x=rep(1:8, 3)
)
aln$mut <- 'no'
aln$mut[c(2,15,20,23)] <- 'yes'
# 生成序列比对图
p2 <- ggplot(aln, aes(x, species)) +
geom_text(aes(label=letter, color=mut, size=mut)) +
scale_x_continuous(breaks=1:10, expand=c(0.105, 0)) +
xlab('') +
scale_color_manual(values=c('black', 'red')) +
scale_size_manual(values=c(5, 6)) +
theme_logo() +
theme(legend.position='none', axis.text.x=element_blank())
# 创建保守性柱状图
bp_data <- data.frame(x=1:8, conservation=sample(1:100, 8))
p3 <- ggplot(bp_data, aes(x, conservation)) +
geom_bar(stat='identity', fill='grey') +
theme_logo() +
scale_x_continuous(breaks=1:10, expand=c(0.105, 0)) +
xlab('')
# 组合图形
cowplot::plot_grid(p1, p2, p3, ncol=1, align='v')
与其他工具集成:
ggseqlogo 可以与其他生物信息学包配合使用,如 ggmotif 包可以直接从 MEME 结果文件中提取 motif 并使用 ggseqlogo 进行可视化,universalmotif 包也提供了与 ggseqlogo 的集成功能。
应用场景
Motif图在基因组学和分子生物学研究中应用广泛:
转录因子结合位点分析:展示DNA序列中转录因子的保守结合模式。
蛋白质结构域分析:显示蛋白质序列中功能结构域的保守氨基酸。
多序列比对可视化:展示多序列比对中的保守区域。
ChIP-seq分析:可视化ChIP-seq实验鉴定出的富集motif。
基因组特征分析:展示基因组特定区域的序列特征。
参考文献
[1] Wagih O. ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics. 2017;33(22):3645-3647. doi:10.1093/bioinformatics/btx469