# 安装包
if (!requireNamespace("ggmsa", quietly = TRUE)) {
install.packages("ggmsa")
}
# 加载包
library(ggmsa)多序列比对可视化
多序列比对(Multiple Sequence Alignment, MSA)是生物信息学中一项基础且关键的技术,用于将三个或更多生物序列(DNA、RNA 或蛋白质)按照其进化或结构上的相似性进行排列,使得同源位点(即来自共同祖先的位点)尽可能对齐。
示例
环境配置
系统要求: 跨平台(Linux/MacOS/Windows)
编程语言:R
依赖包:
ggmsa
sessioninfo::session_info("attached")─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.6.0 (2026-04-24)
os Ubuntu 24.04.4 LTS
system x86_64, linux-gnu
ui X11
language (EN)
collate C.UTF-8
ctype C.UTF-8
tz UTC
date 2026-05-09
pandoc 3.1.3 @ /usr/bin/ (via rmarkdown)
quarto 1.9.37 @ /usr/local/bin/quarto
─ Packages ───────────────────────────────────────────────────────────────────
package * version date (UTC) lib source
ggmsa * 1.18.0 2026-04-28 [1] Bioconduc~
[1] /home/runner/work/_temp/Library
[2] /opt/R/4.6.0/lib/R/site-library
[3] /opt/R/4.6.0/lib/R/library
* ── Packages attached to the search path.
──────────────────────────────────────────────────────────────────────────────数据准备
DNA/RNA/氨基酸序列数据通常以FASTA格式存储,每个序列由一个描述行(以“>”开头)和随后的序列行组成。
# 示例数据
protein_fasta <- system.file("extdata", "sample.fasta", package = "ggmsa")
# 查看数据
seqs <- readLines(protein_fasta)
head(seqs)[1] ">PH4H_Rattus_norvegicus"
[2] "MAAVVLENGVLSRKLSDFGQETSYIEDNSNQNGAISLIFSLKEEVGALAKVLRLFEENDINLTHIESRPSRLNKDEYEFF"
[3] "TYLDKRTKPVLGSIIKSLRNDIGATVHELSRDKEKNTVPWFPRTIQELDRFANQILSYGAELDADHPGFKDPVYRARRKQ"
[4] "FADIAYNYRHGQPIPRVEYTEEEKQTWGTVFRTLKALYKTHACYEHNHIFPLLEKYCGFREDNIPQLEDVSQFLQTCTGF"
[5] "RLRPVAGLLSSRDFLGGLAFRVFHCTQYIRHGSKPMYTPEPDICHELLGHVPLFSDRSFAQFSQEIG-LASLGAPDEYIE"
[6] "KLATIYWFTVEFGLCKEG-DSIKAYGAGLLSSFGELQYCLSD-KPKLLPLELEKTACQEYSVTEFQPLYYVAESFSDAKE"可视化
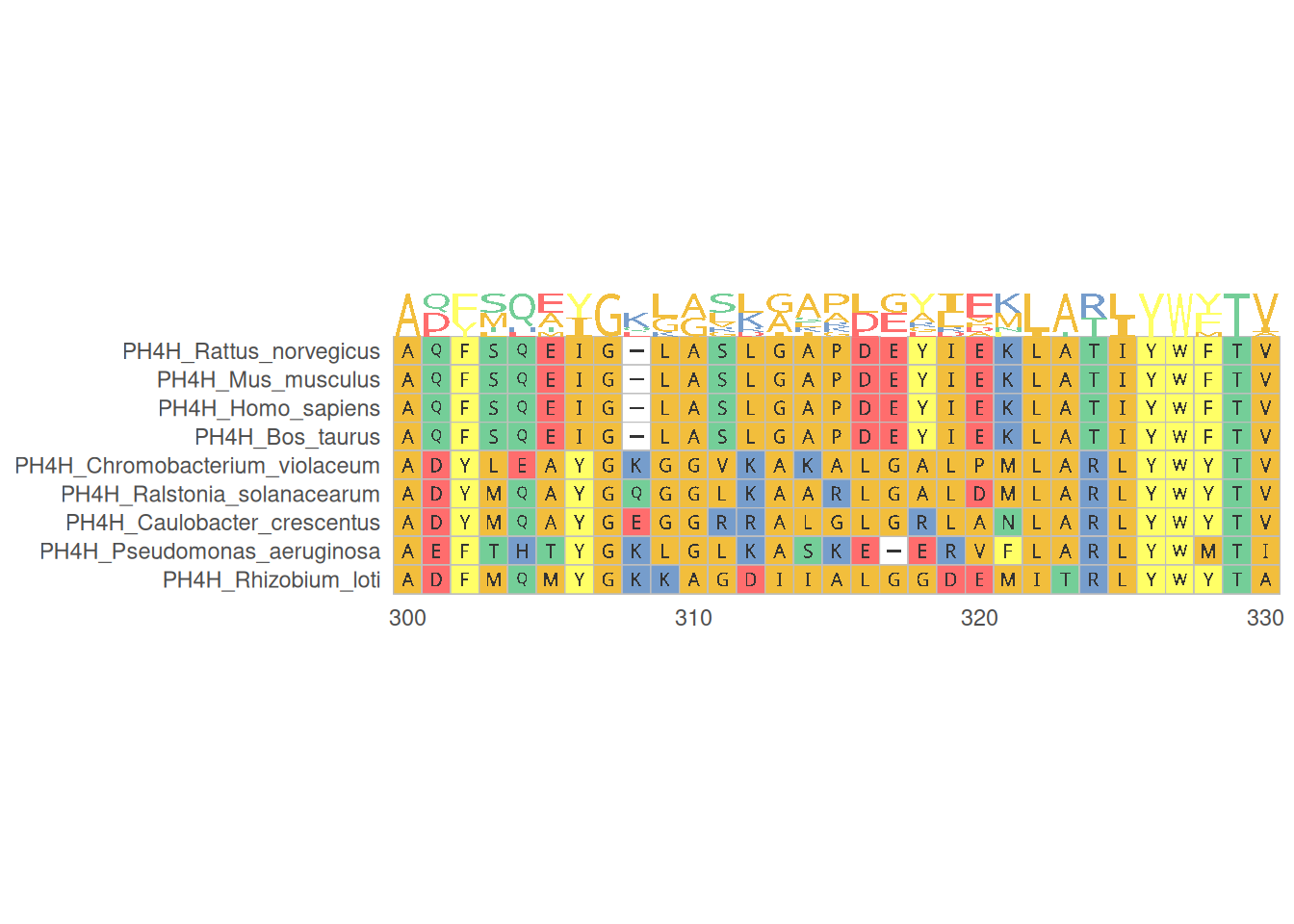
1. 蛋白质多序列比对
蛋白质多序列比对可视化展示了多个蛋白质序列在特定区域的比对情况,帮助识别保守区域和变异位点,提供专业的氨基酸序列颜色方案。
# 蛋白质多序列比对
p <- ggmsa(
protein_fasta,
start = 300,
end = 330,
font = "DroidSansMono",
color = "Chemistry_AA",
char_width = 0.5,
seq_name = TRUE,
consensus_views = FALSE
)
p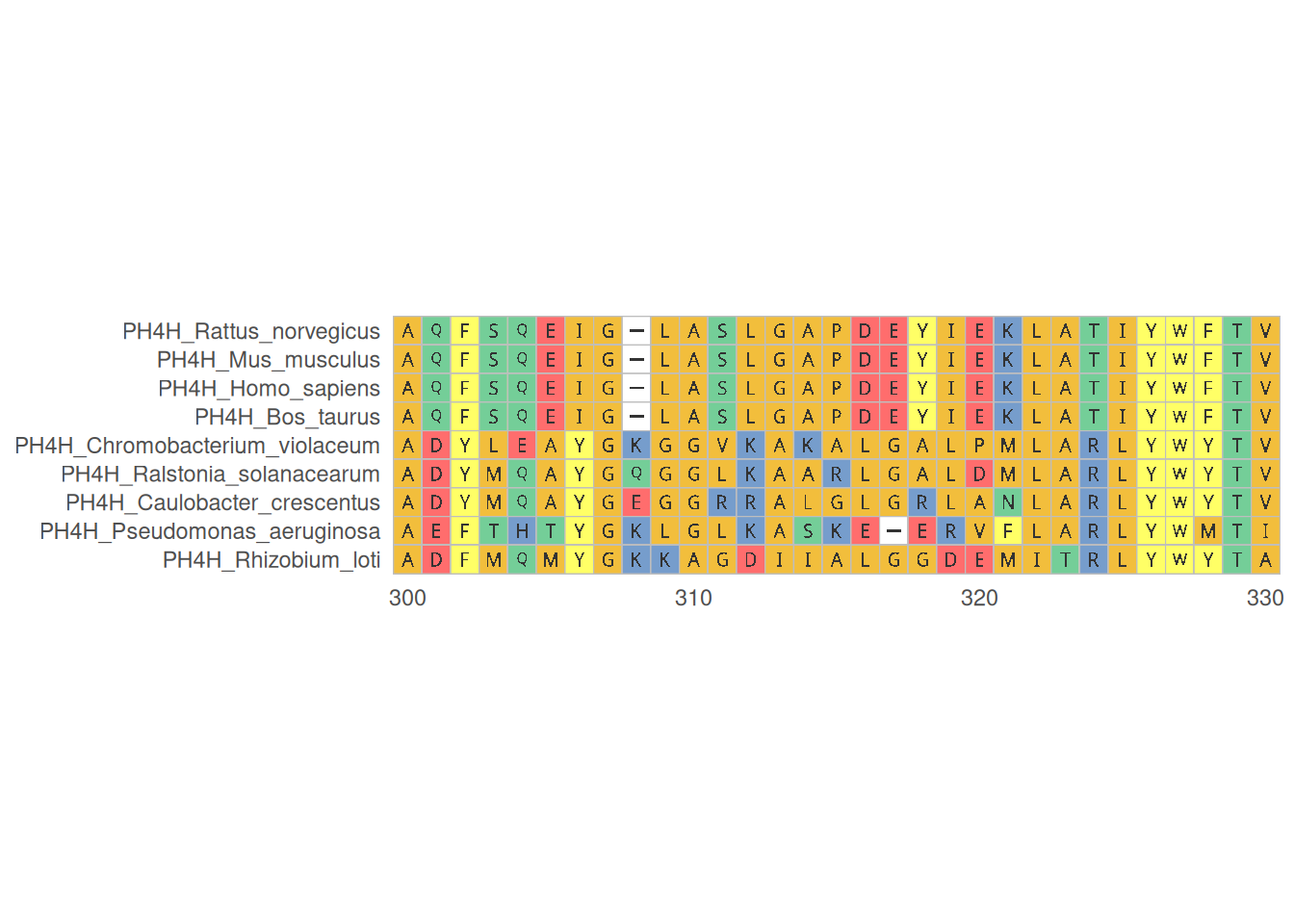
2. 多序列比对及统计
多序列比对结果统计图展示了多个序列在比对区域内的保守性和变异情况,帮助识别关键位点和区域。
# 多序列比对及统计
p <- ggmsa(
protein_fasta,
start = 300,
end = 320,
font = "DroidSansMono",
color = "Chemistry_AA",
char_width = 0.5,
seq_name = TRUE,
consensus_views = FALSE
) +
geom_msaBar()
p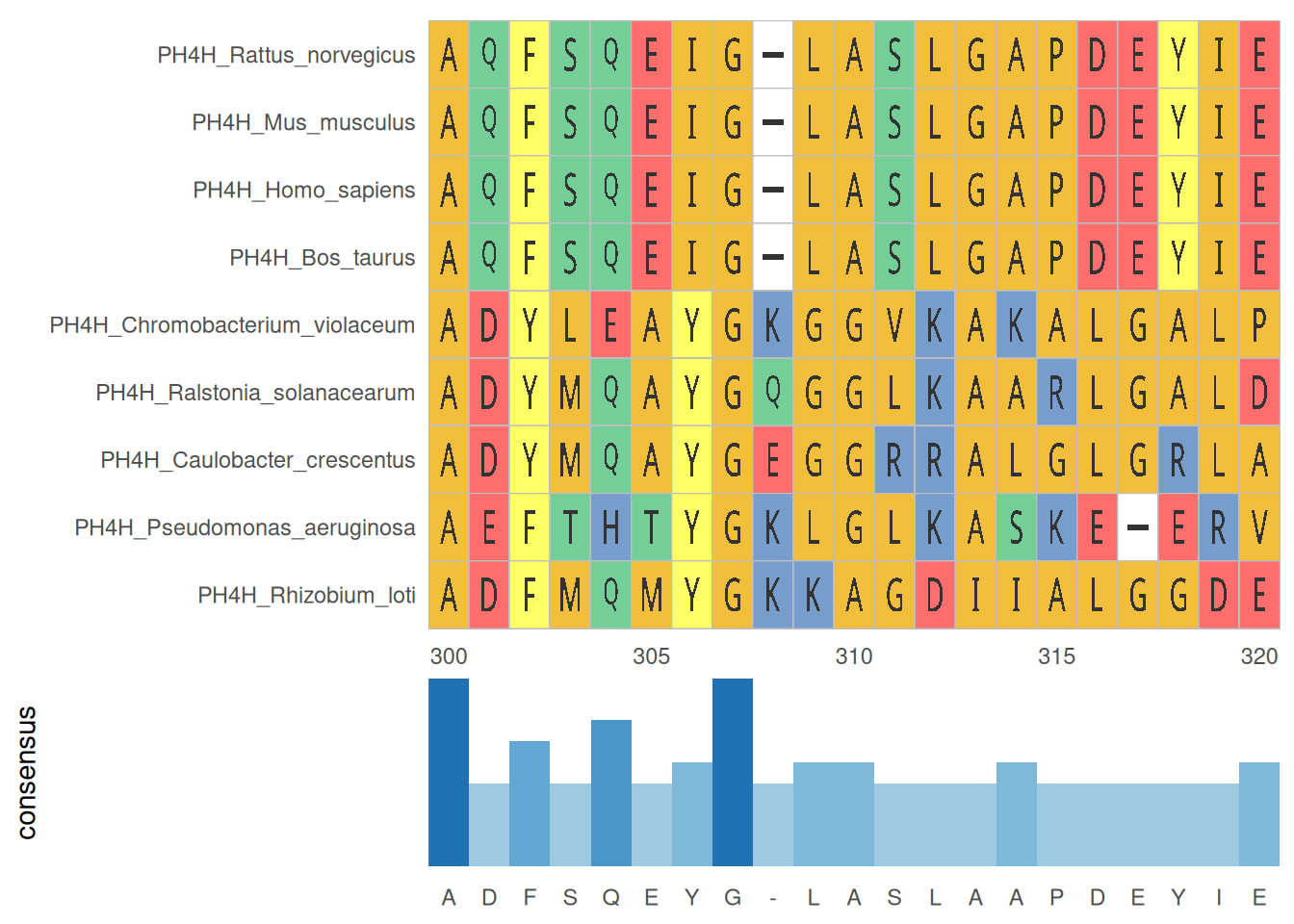
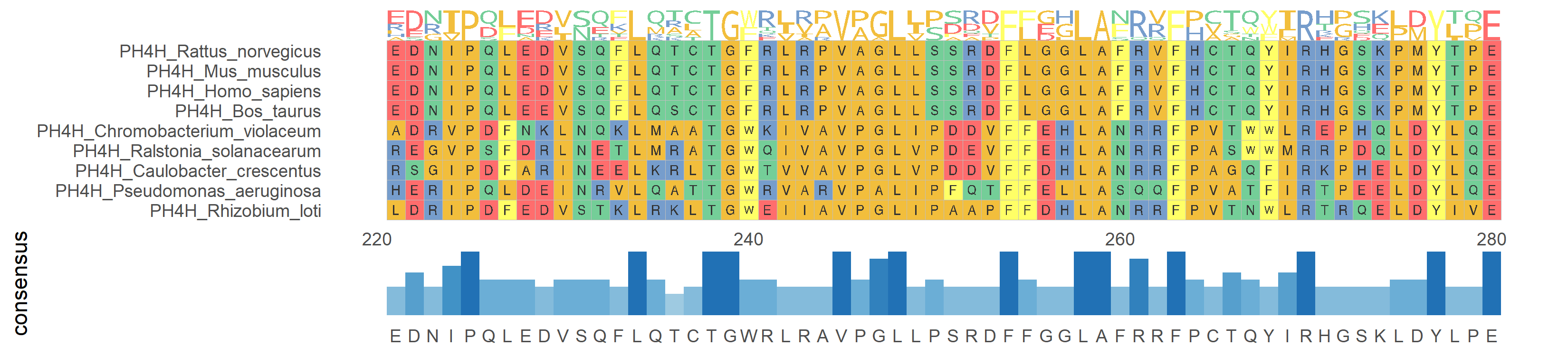
3. 多序列比对及Logo
多序列比对及Logo展示比对一致性结果的同时,对多个序列的碱基或氨基酸进行排序以Logo形式显示。
# 多序列比对及Logo
p <- ggmsa(
protein_fasta,
start = 300,
end = 330,
font = "DroidSansMono",
color = "Chemistry_AA",
char_width = 0.5,
seq_name = TRUE,
consensus_views = FALSE
) +
geom_seqlogo()
p