# 安装包
if (!requireNamespace("CMplot", quietly = TRUE)) {
install.packages("CMplot")
}
# 加载包
library(CMplot)全基因组关联分析圈图
全基因组关联分析(GWAS)结果可视化主要包含按染色体位置展示SNP圈图、SNP密度图、显著性筛选的曼哈顿图、比较观测 p 值与期望 p 值的分布的QQ图等,用于筛选到全基因水平的候选变异基因。
示例

环境配置
系统要求: 跨平台(Linux/MacOS/Windows)
编程语言:R
依赖包:
CMplot
sessioninfo::session_info("attached")─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.6.0 (2026-04-24)
os Ubuntu 24.04.4 LTS
system x86_64, linux-gnu
ui X11
language (EN)
collate C.UTF-8
ctype C.UTF-8
tz UTC
date 2026-05-09
pandoc 3.1.3 @ /usr/bin/ (via rmarkdown)
quarto 1.9.37 @ /usr/local/bin/quarto
─ Packages ───────────────────────────────────────────────────────────────────
package * version date (UTC) lib source
CMplot * 4.5.1 2024-01-19 [1] RSPM
[1] /home/runner/work/_temp/Library
[2] /opt/R/4.6.0/lib/R/site-library
[3] /opt/R/4.6.0/lib/R/library
* ── Packages attached to the search path.
──────────────────────────────────────────────────────────────────────────────数据准备
示例数据包含60K液相芯片Pig的原始变异信息数据。
# 示例数据
data(pig60K)
data <- pig60K
# 查看数据
head(data, 5) SNP Chromosome Position trait1 trait2 trait3
1 ALGA0000009 1 52297 0.7738187 0.5119432 0.5119432
2 ALGA0000014 1 79763 0.7738187 0.5119432 0.5119432
3 ALGA0000021 1 209568 0.7583016 0.9840529 0.9840529
4 ALGA0000022 1 292758 0.7200305 0.4888714 0.4888714
5 ALGA0000046 1 747831 0.9736840 0.2209684 0.2209684可视化
1. SNP筛选基因组圈图
基因组圈图可以在相同占比下展示更多的染色体和性状,在多个性状比较时占极大优势。
# SNP筛选基因组圈图
p <- CMplot(
data,
type = "p",
plot.type = "c",
chr.labels = paste("Chr", c(1:18, "X", "Y"), sep = ""),
r = 8,
cir.axis = TRUE,
outward = TRUE,
cir.axis.col = "black",
cir.chr.h = 2,
chr.den.col = "black",
file.output = FALSE,
verbose = FALSE,
mar = c(0,0,0,0)
)
pNULL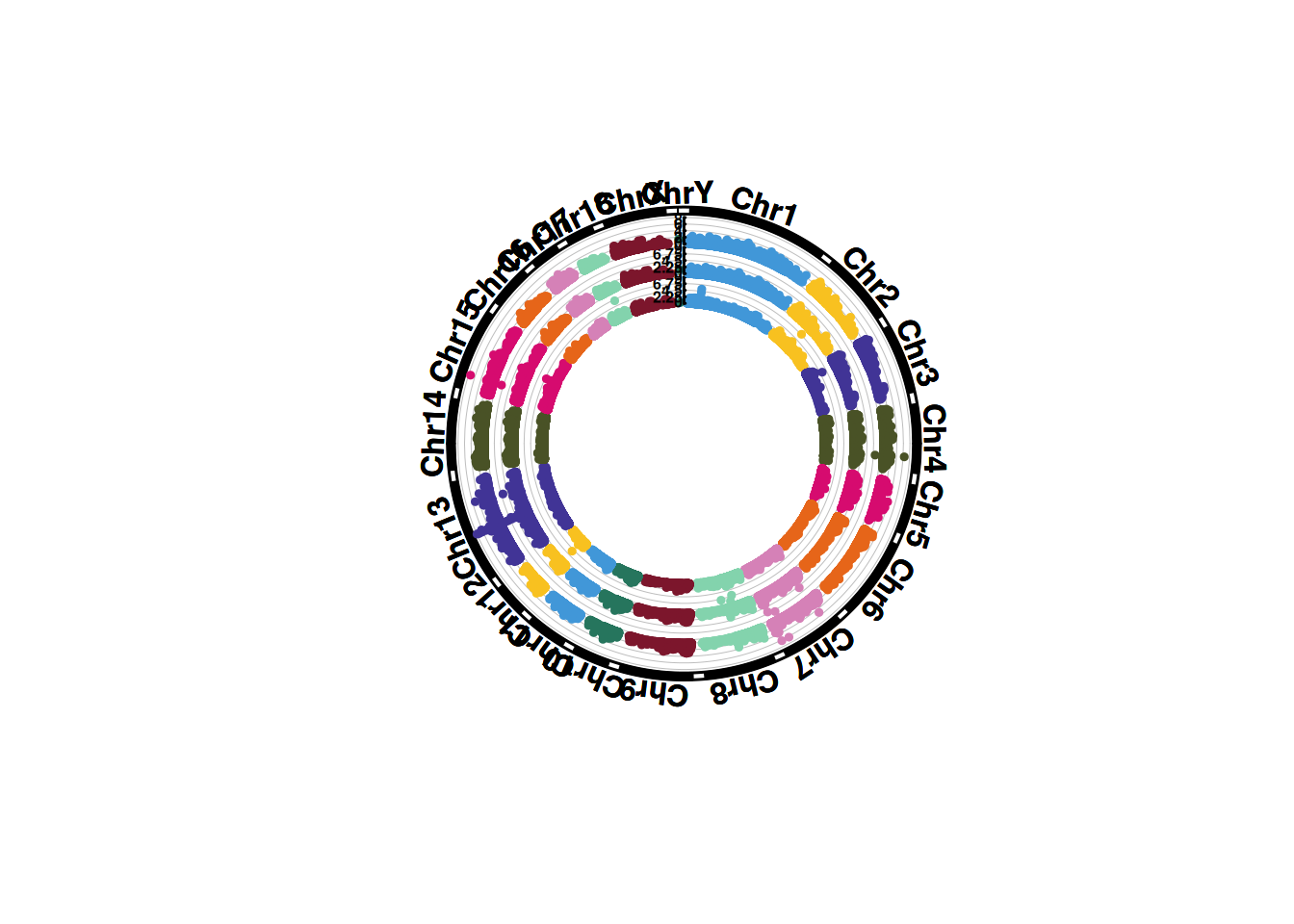
2. SNP染色体密度图
SNP染色体密度图展示各个染色体上SNP密度分布。
# SNP染色体密度图
p <- capture.output(
CMplot(
data,
plot.type = "d",
bin.size = 1e6,
chr.den.col = c("darkgreen", "yellow", "red"),
main = "Density",
file.output = FALSE,
verbose = FALSE
)
)
print(p[1])[1] NA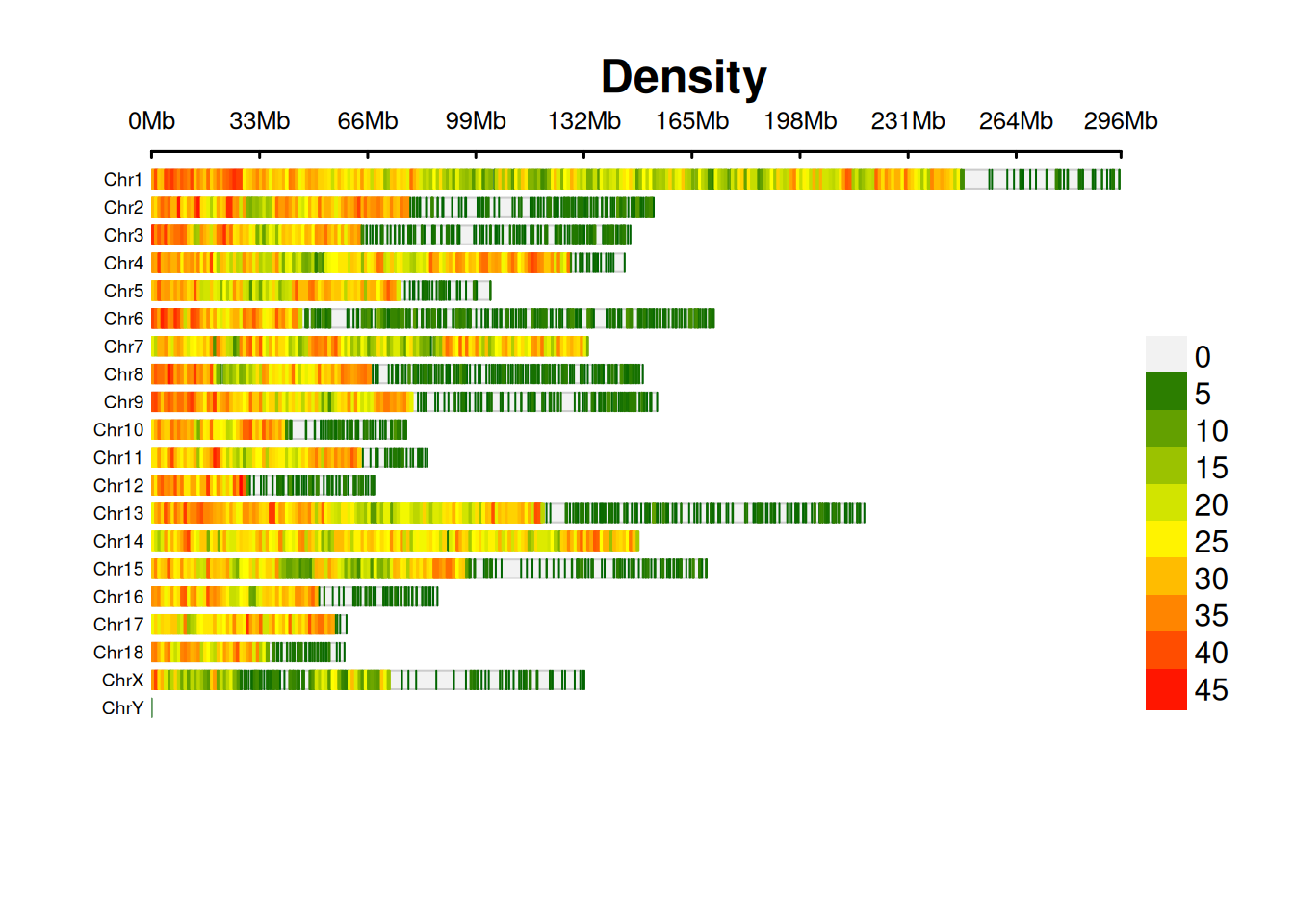
3. SNP筛选曼哈顿图
SNP筛选曼哈顿图便于展示5%或者1%的SNP显著性,同时也辅助鉴定关键染色体的集中效应。
# SNP筛选曼哈顿图
p <- CMplot(
data,
type = "p",
plot.type = "m",
LOG10 = TRUE,
threshold = NULL,
file.output = FALSE,
verbose = FALSE,
chr.labels.angle = 45
)
pNULL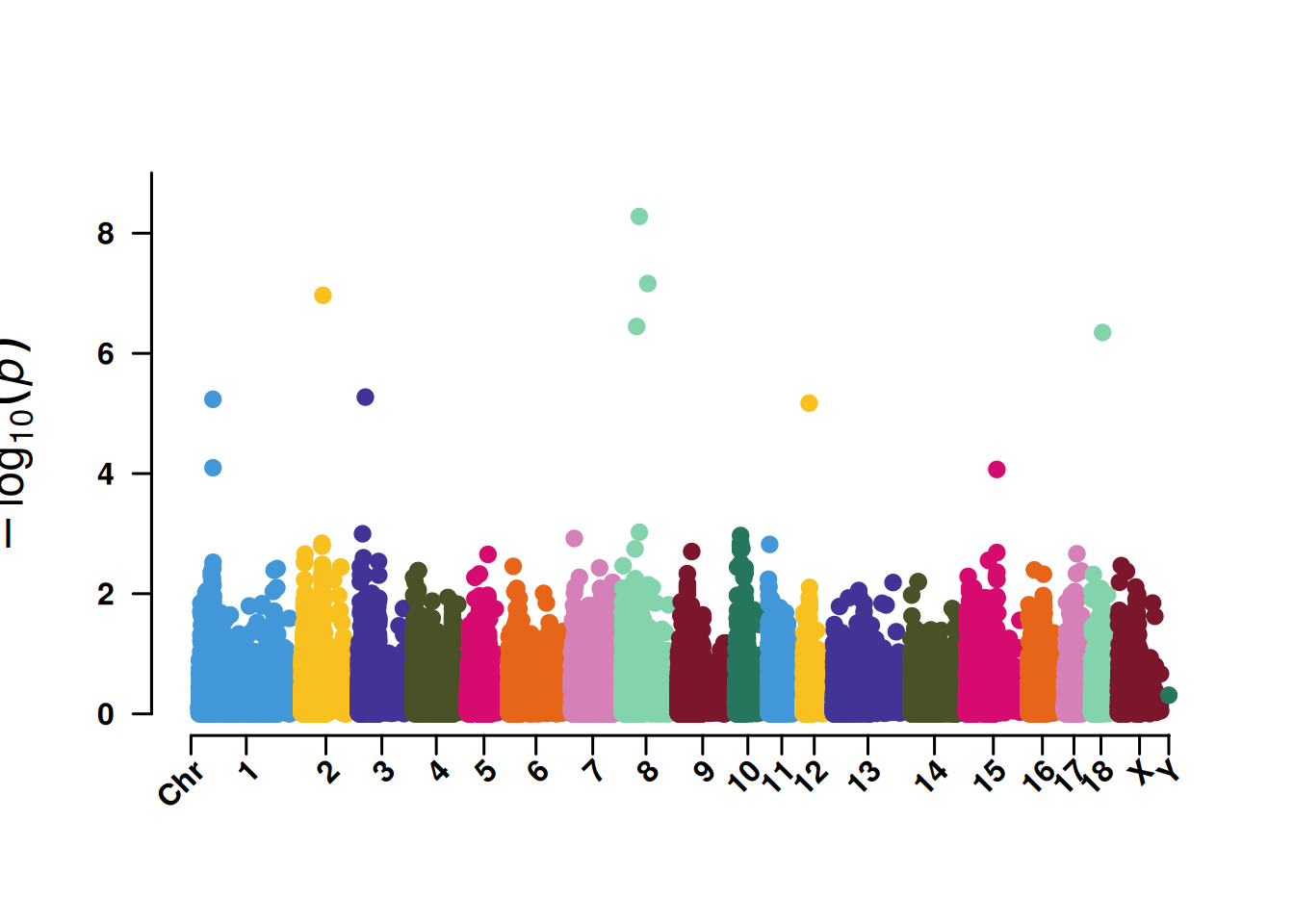
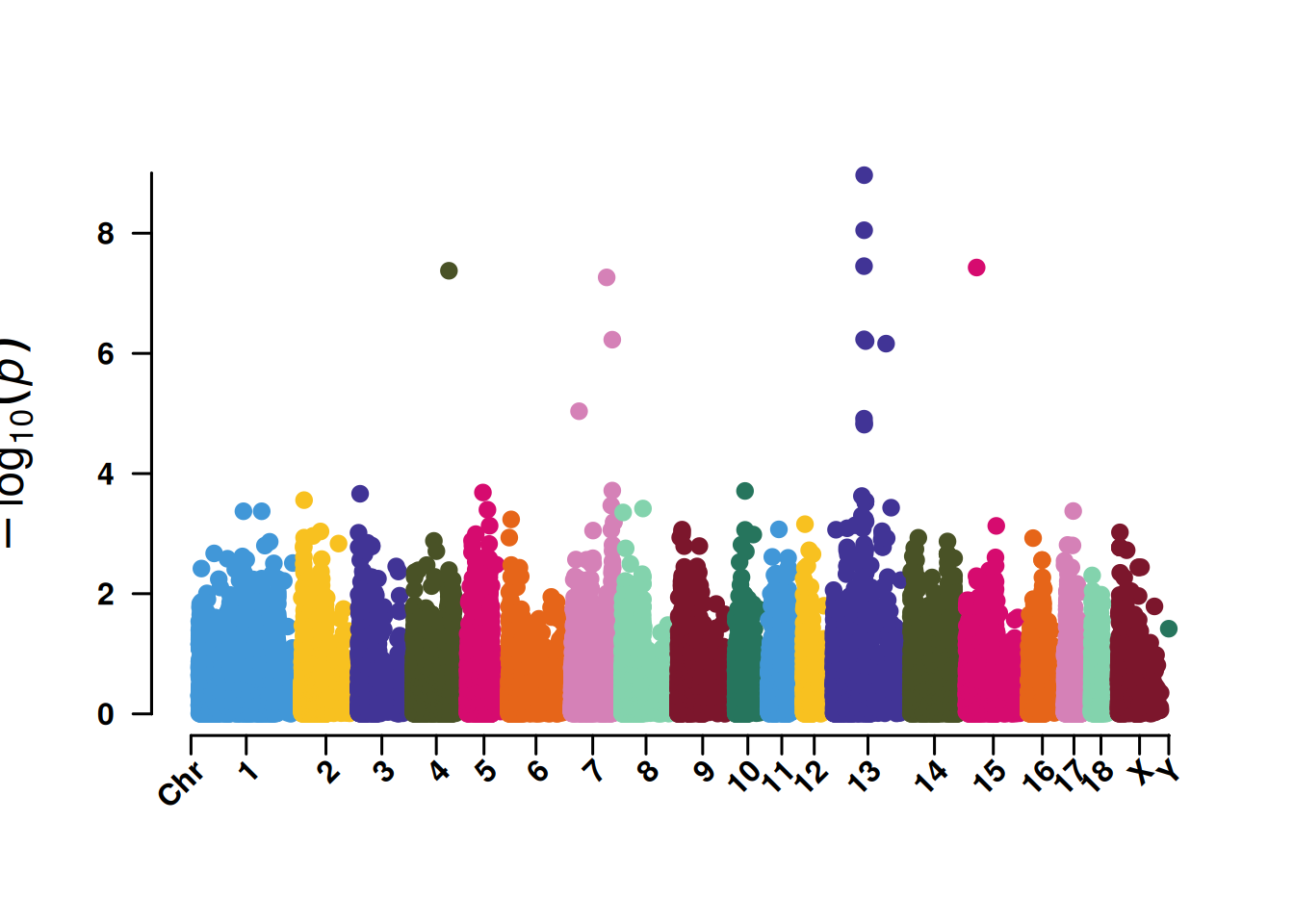
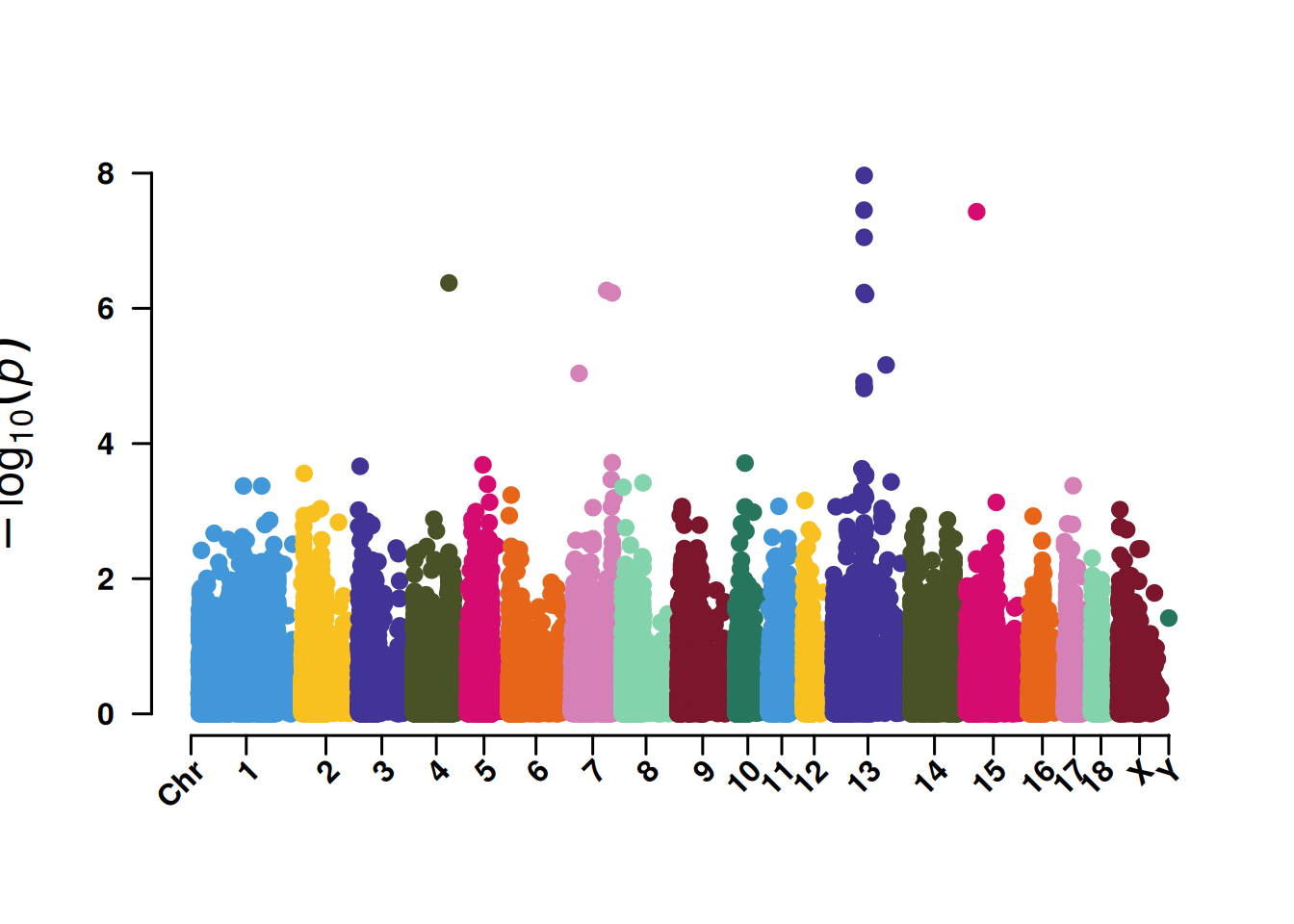
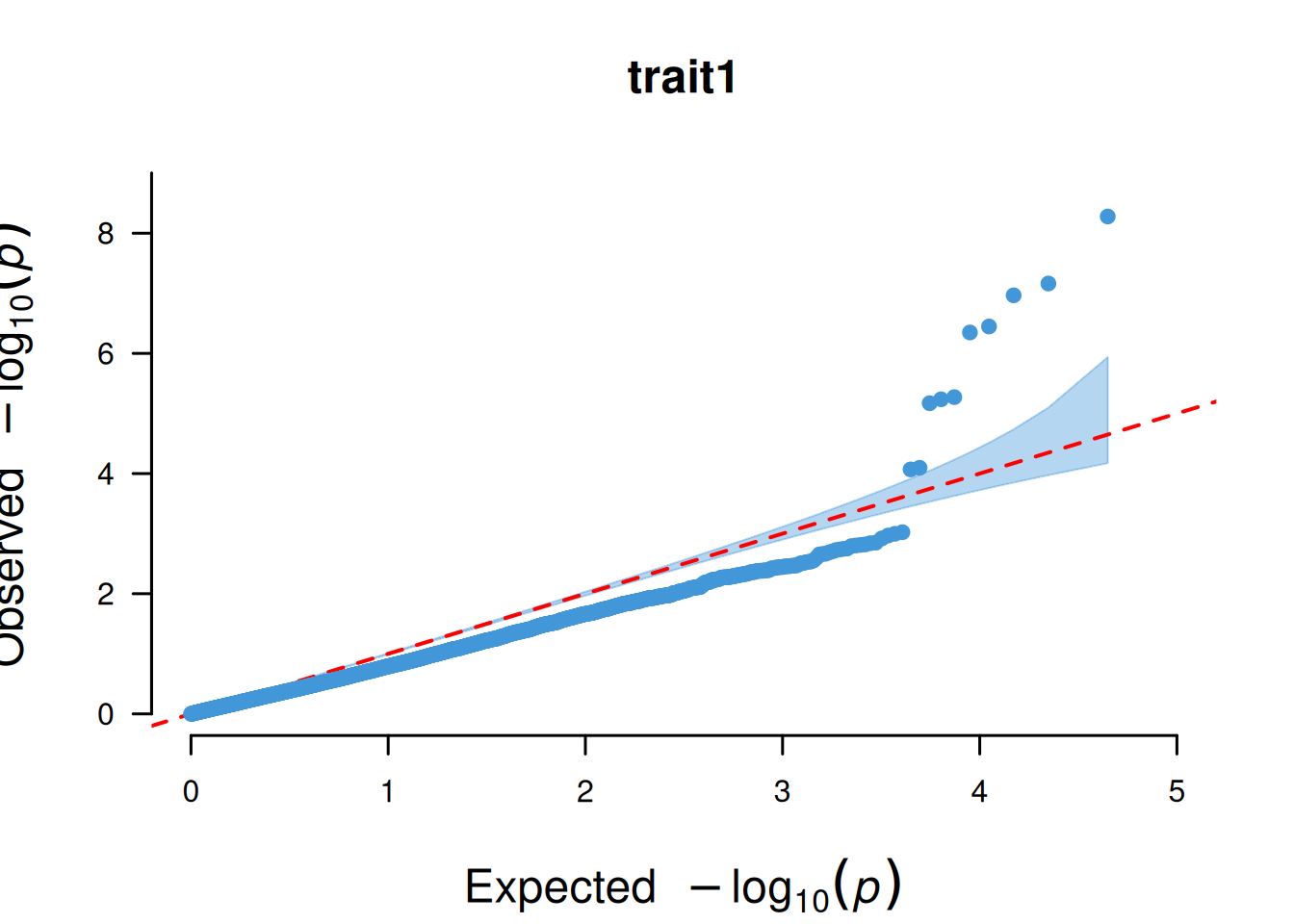
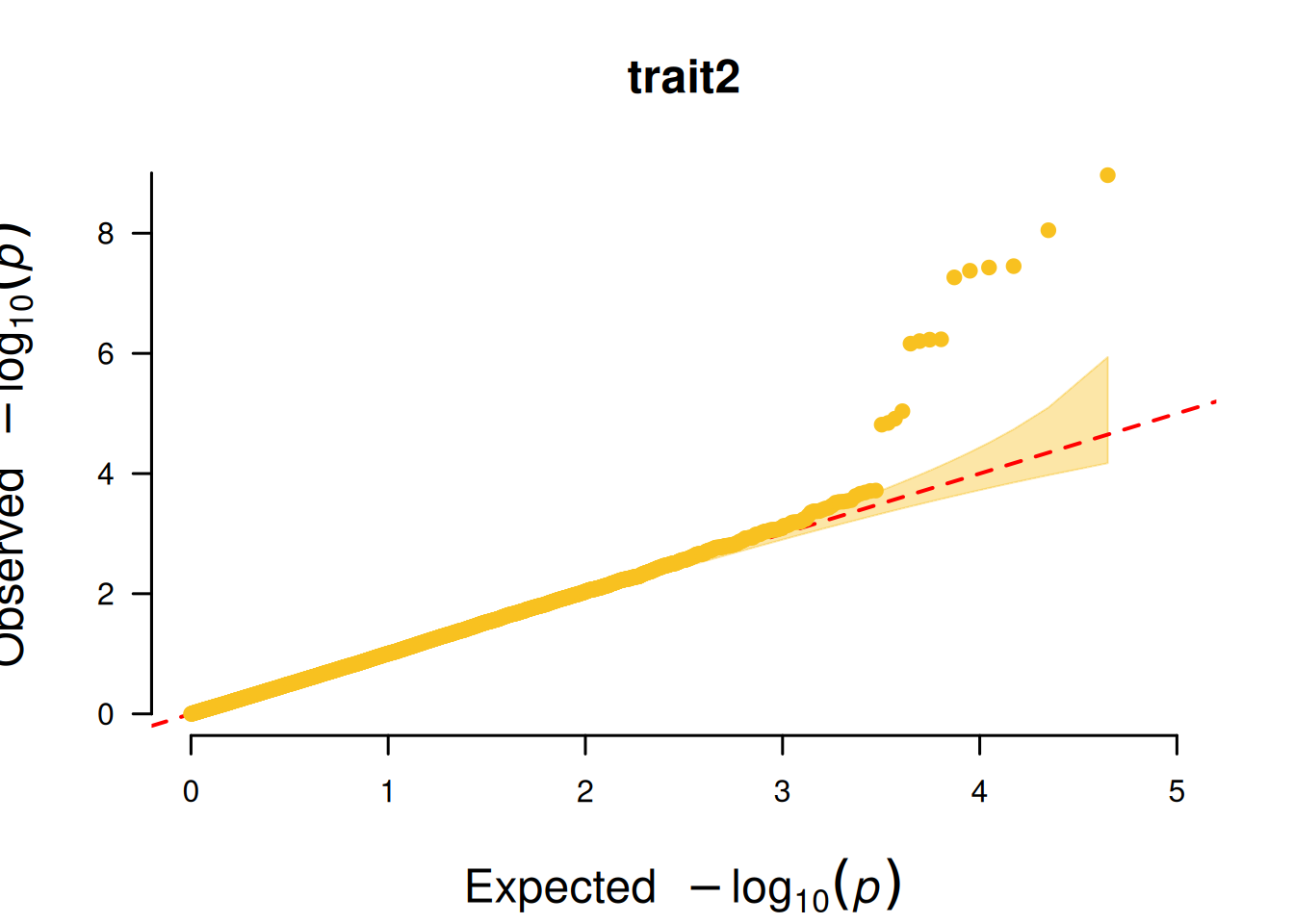
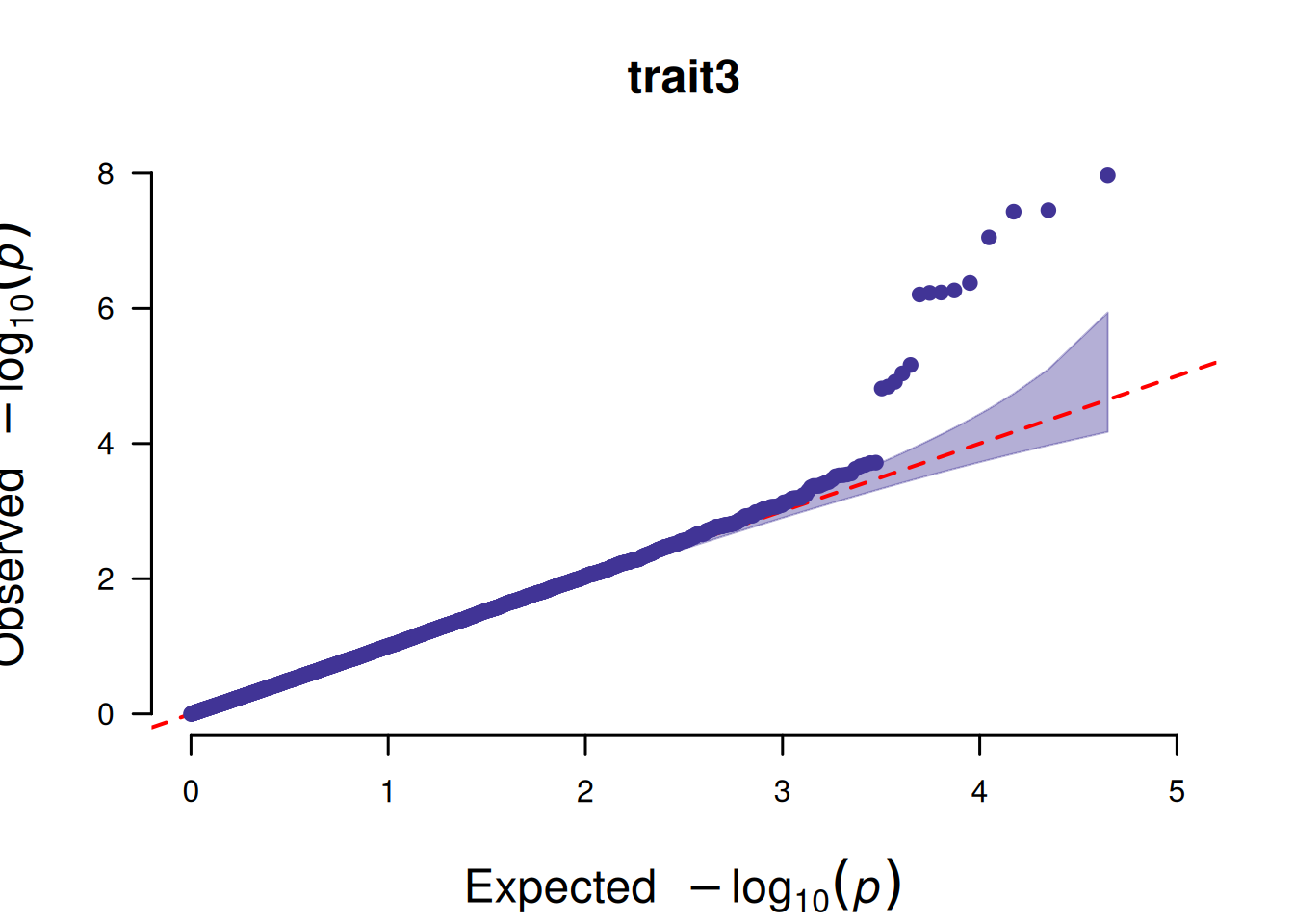
4. SNP基于的QQ图
SNP基于的QQ图核心作用是检验 GWAS 分析中 SNP 的 P 值分布是否符合 “无关联假设下的期望分布”,以此判断分析结果的可靠性。
# SNP基于的QQ图
p <- CMplot(
data,
plot.type = "q",
box = FALSE,
conf.int = TRUE,
conf.int.col = NULL,
threshold.col = "red",
threshold.lty = 2,
file.output = FALSE,
verbose = FALSE
)
pNULL