# 安装包
if (!requireNamespace("pathview", quietly = TRUE)) {
remotes::install_github("datapplab/pathview")
}
if (!requireNamespace("dbplyr", quietly = TRUE)) {
install.packages("dbplyr")
}
# 加载包
library(pathview)
library(dbplyr)KEGG通路图
示例
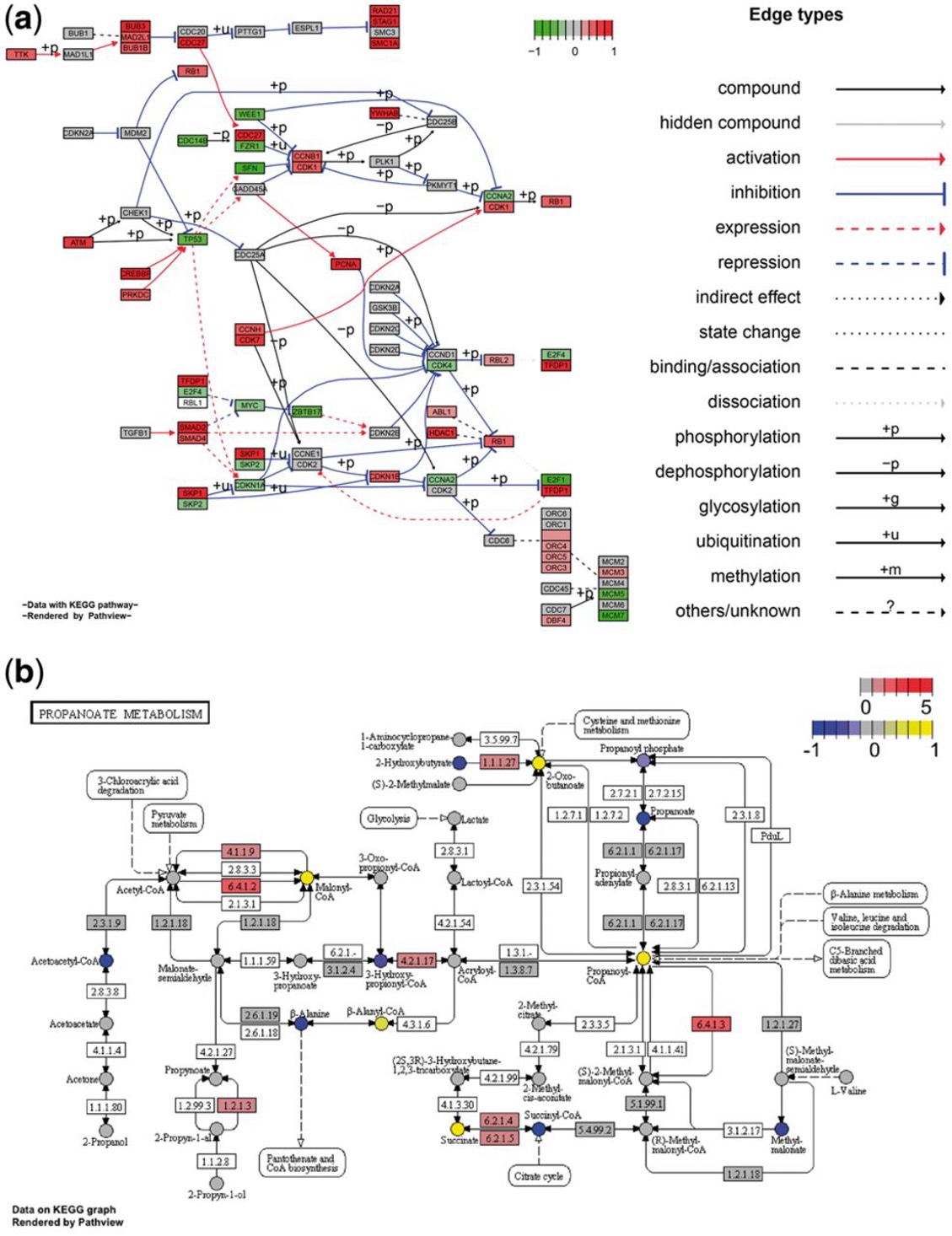
(a)展示了包含基因数据的经典信号通路(hsa04110 Cell cycle)的Graphviz视图;(b)展示了包含离散基因数据和连续代谢物数据的代谢通路(hsa00640 Propanoate metabolism)的KEGG视图。
图片、R包等均来源于Luo等人的文章。[1]
环境配置
系统要求: 跨平台(Linux/MacOS/Windows)
编程语言:R
依赖包:
pathview,dbplyr
sessioninfo::session_info("attached")─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.6.0 (2026-04-24)
os Ubuntu 24.04.4 LTS
system x86_64, linux-gnu
ui X11
language (EN)
collate C.UTF-8
ctype C.UTF-8
tz UTC
date 2026-05-09
pandoc 3.1.3 @ /usr/bin/ (via rmarkdown)
quarto 1.9.37 @ /usr/local/bin/quarto
─ Packages ───────────────────────────────────────────────────────────────────
package * version date (UTC) lib source
dbplyr * 2.5.2 2026-02-13 [1] RSPM
pathview * 1.47.1 2026-05-04 [1] Github (datapplab/pathview@1f17eb7)
[1] /home/runner/work/_temp/Library
[2] /opt/R/4.6.0/lib/R/site-library
[3] /opt/R/4.6.0/lib/R/library
* ── Packages attached to the search path.
──────────────────────────────────────────────────────────────────────────────数据准备
示例数据为基因表达矩阵: 行为基因的ENTREZ ID,列为样本名,表达数据经过归一化处理。其实这里可以为任何数值型数据,例如fold change, p-value等等,根据自己的需求来定。
data("gse16873.d")
head(gse16873.d) DCIS_1 DCIS_2 DCIS_3 DCIS_4 DCIS_5
10000 -0.30764480 -0.14722769 -0.023784808 -0.07056193 -0.001323087
10001 0.41586805 -0.33477259 -0.513136907 -0.16653712 0.111122223
10002 0.19854925 0.03789588 0.341865341 -0.08527420 0.767559264
10003 -0.23155297 -0.09659311 -0.104727283 -0.04801404 -0.208056443
100048912 -0.04490724 -0.05203146 0.036390376 0.04807823 0.027205816
10004 -0.08756237 -0.05027725 0.001821133 0.03023835 0.008034394
DCIS_6
10000 -0.15026813
10001 0.13400734
10002 0.15828609
10003 0.03344448
100048912 0.05444739
10004 -0.06860749gene_data <- as.data.frame(gse16873.d)
head(gene_data) DCIS_1 DCIS_2 DCIS_3 DCIS_4 DCIS_5
10000 -0.30764480 -0.14722769 -0.023784808 -0.07056193 -0.001323087
10001 0.41586805 -0.33477259 -0.513136907 -0.16653712 0.111122223
10002 0.19854925 0.03789588 0.341865341 -0.08527420 0.767559264
10003 -0.23155297 -0.09659311 -0.104727283 -0.04801404 -0.208056443
100048912 -0.04490724 -0.05203146 0.036390376 0.04807823 0.027205816
10004 -0.08756237 -0.05027725 0.001821133 0.03023835 0.008034394
DCIS_6
10000 -0.15026813
10001 0.13400734
10002 0.15828609
10003 0.03344448
100048912 0.05444739
10004 -0.06860749可视化
1. 基础绘图
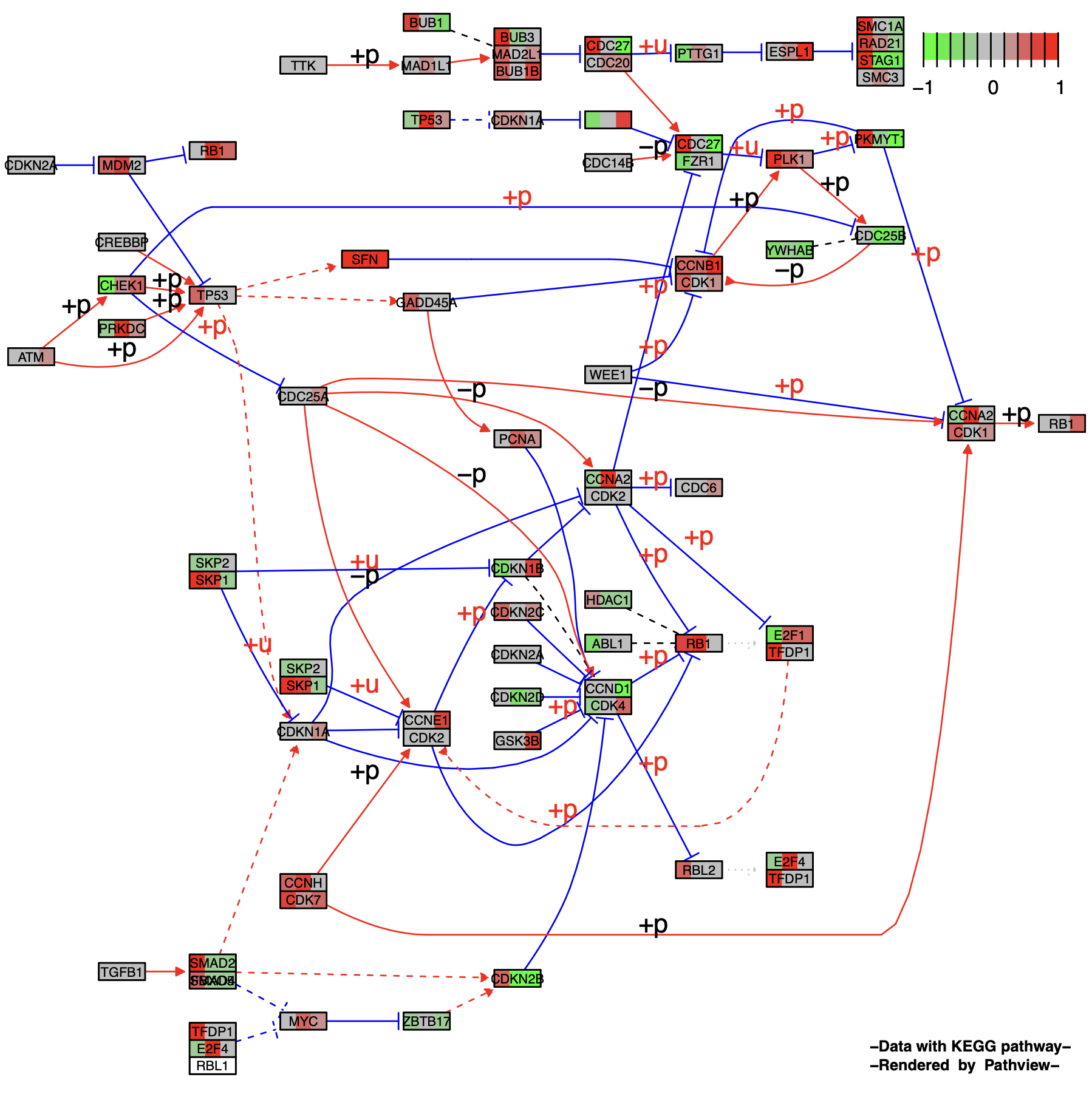
观察单个样本(DCIS_1)的细胞周期信号通路图(KEGG ID: 04110)。
1.1 KEGG视图
当设置kegg.native = TRUE时,输出的通路图是基于原始KEGG视图绘制的。输出图像自动保存在了当前目录下。
p1 <- pathview(gene.data = gse16873.d[, 1], # 输入基因矩阵
pathway.id = "04110", # 通路ID
species = "hsa", # 物种:人类
out.suffix = "gse16873_KEGG", # 输出文件的后缀
kegg.native = T, # 以原始KEGG视图输出
same.layer = T # 绘制单个图层
)
p1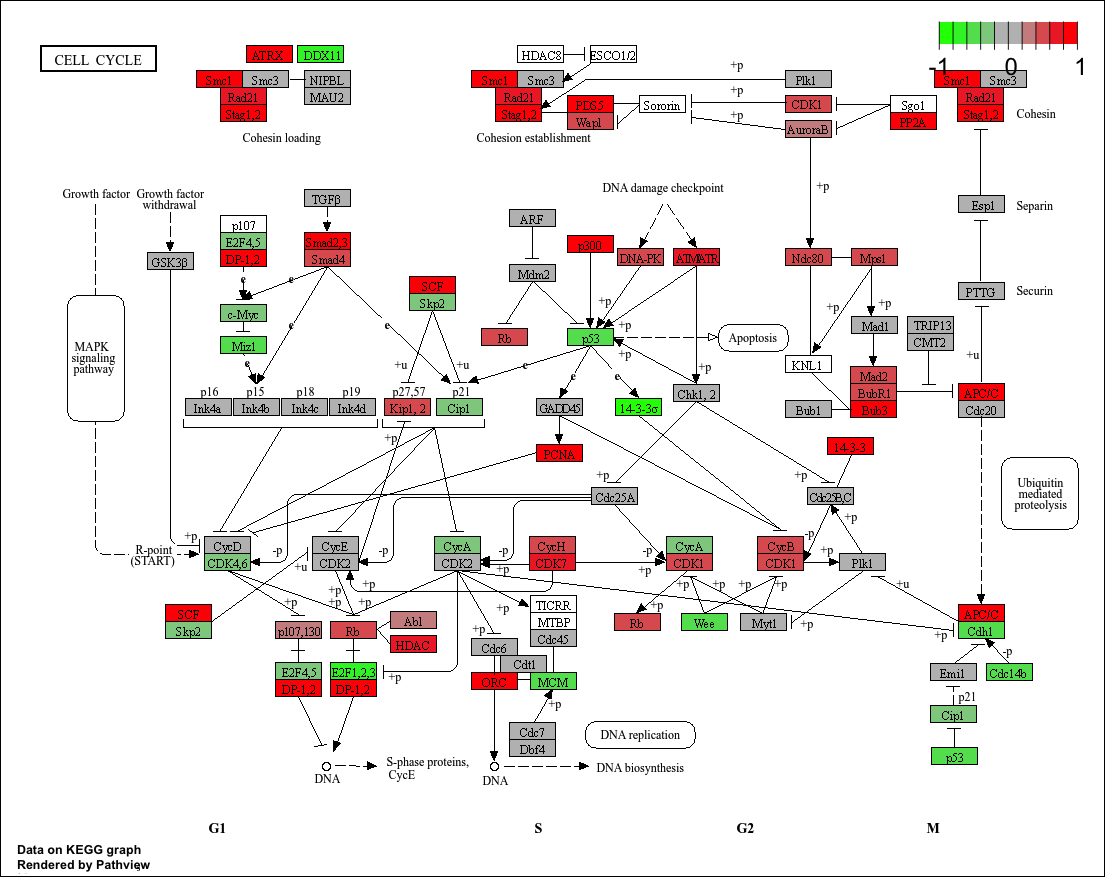
为了更好的理解kegg.native = TRUE,我们对比一下KEGG官网的Cell cycle的通路图(https://www.kegg.jp/pathway/map04110)
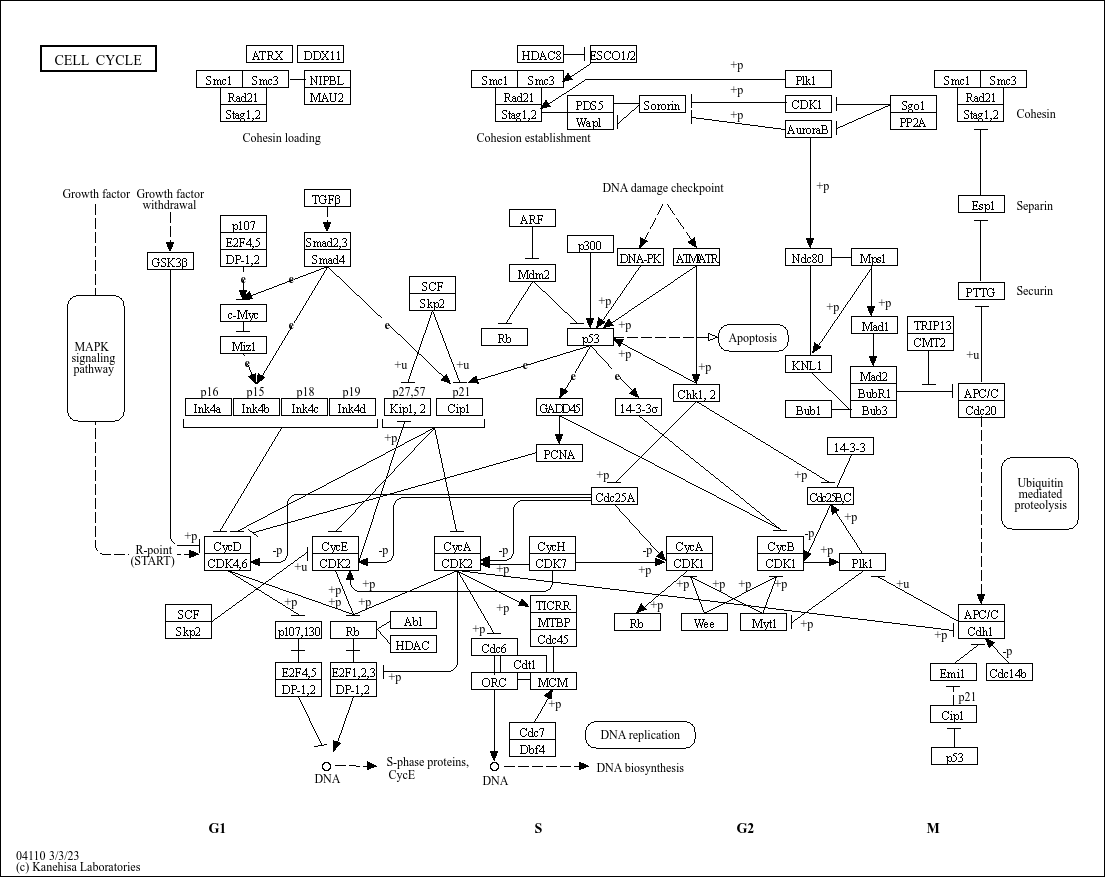
不难发现,我们输出的结果图其实就是在官网提供的通路图的基础上,对研究样本的通路基因的表达差异进行了可视化。而为当kegg.native = FALSE时,则改为使用Graphviz引擎制图,输出是PDF格式的向量图,这个后续会介绍。
然后我们再查看一下pathview函数输出结果p1的结构,其为一个长度为2的list。包含了我们绘图所需要的数据。
提示
plot.data.gene (data.frame):储存画图用到的基因数据plot.data.cpd (data.frame):储存画图用到的化合物数据kegg.names: 映射节点的基因/化合物的KEGG格式ID(即 Entrez Gene ID 或 KEGG Compound Accessions)labels: 映射节点的基因/化合物的名称all.mapped: 映射到该节点的所有基因/化合物的ID。type: 节点类型。(“基因”、“酶”、“化合物”、“同源物”)x: 原始KEGG通路图中的x坐标。y: 原始KEGG通路图中的y坐标。width: 原始KEGG通路图中的节点宽度。height: 原始KEGG通路图中的节点高度。mol.data: 映射节点的基因/化合物的数据(对应输入数据gene.data)mol.data: 映射节点的基因/化合物的颜色代码
1.2 Graphviz视图
介绍了以KEGG原始形式输出图像(kegg.native = TRUE)后,我们来看下使用Graphviz输出的图(kegg.native = FALSE)的图像格式。
p2 <- pathview(gene.data = gse16873.d[, 1],
pathway.id = "04110",
species = "hsa",
out.suffix = "gse16873_Graph",
kegg.native = F ,# 以Graphviz输出形式输出
same.layer = T # 绘制单个图层
)
p2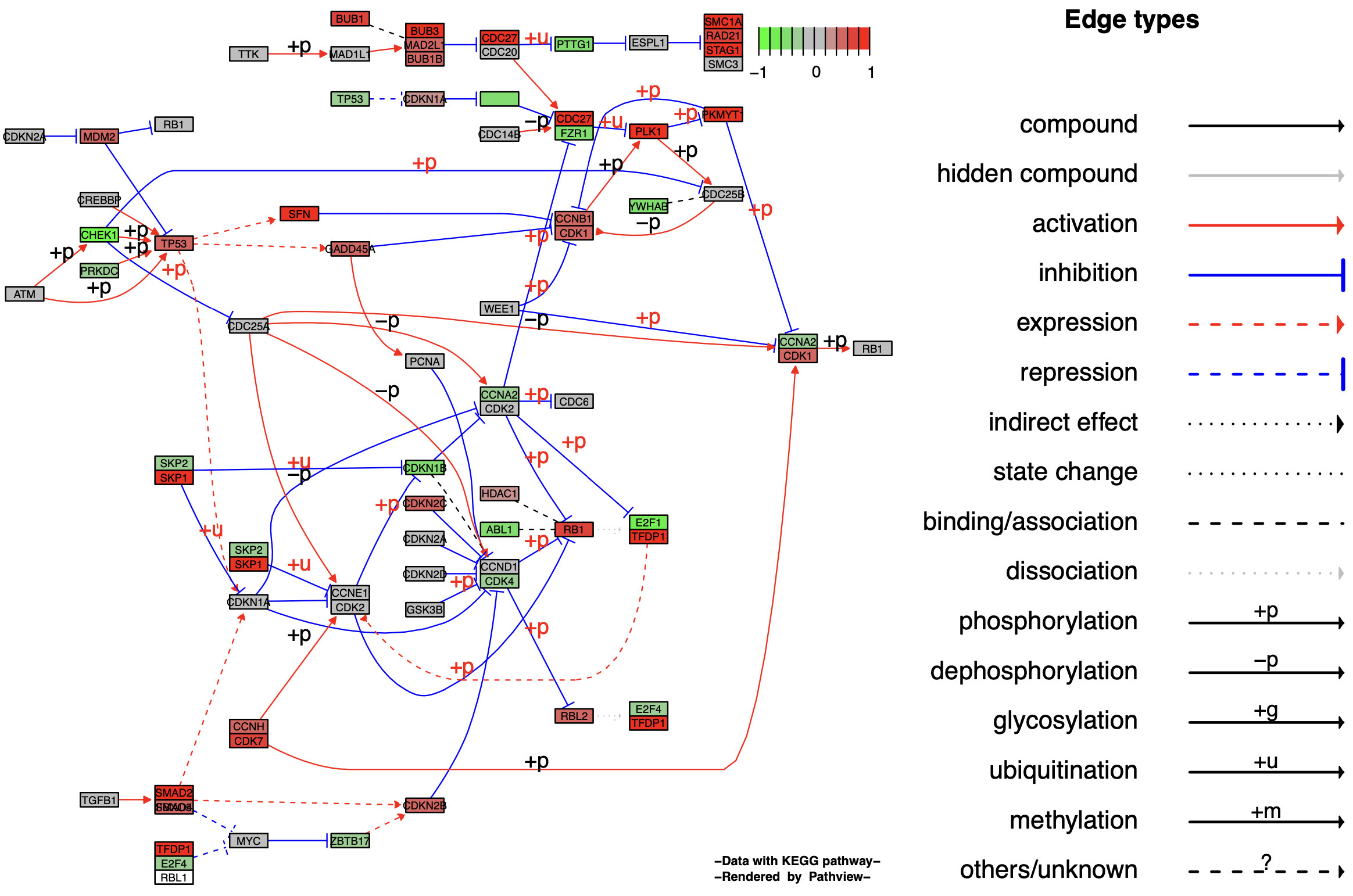
KEGG原始格式图保留了所有通路的元数据,即空间和时间信息、组织/细胞类型、输入、输出和连接,这对通路生物学的可读性和解释很重要。而Graphviz视图为PDF格式矢量图,可以更好地控制节点和边缘属性,更好地查看路径拓扑,如能够用Adobe illustration进行进一步美化。
在Graphviz形式输出的通路图中,same.layer参数的意义与KEGG原始图有所不同,转为描述图例与通路图的图层关系。当same.layer = TRUE时,图例和通路图处于同一个图层之中,并且图例只描述了边缘类型(Egde types),缺少了节点类型(Node types)。
当same.layer = FALSE时,图例和通路图分别处于两个图层,图例同时描述了边缘类型(Egde types)和节点类型(Node types)。
p3 <- pathview(gene.data = gse16873.d[, 1],
pathway.id = "04110",
species = "hsa",
out.suffix = "gse16873_Graph",
kegg.native = F ,# 以Graphviz输出形式输出
same.layer = T # 绘制单个图层
)
p3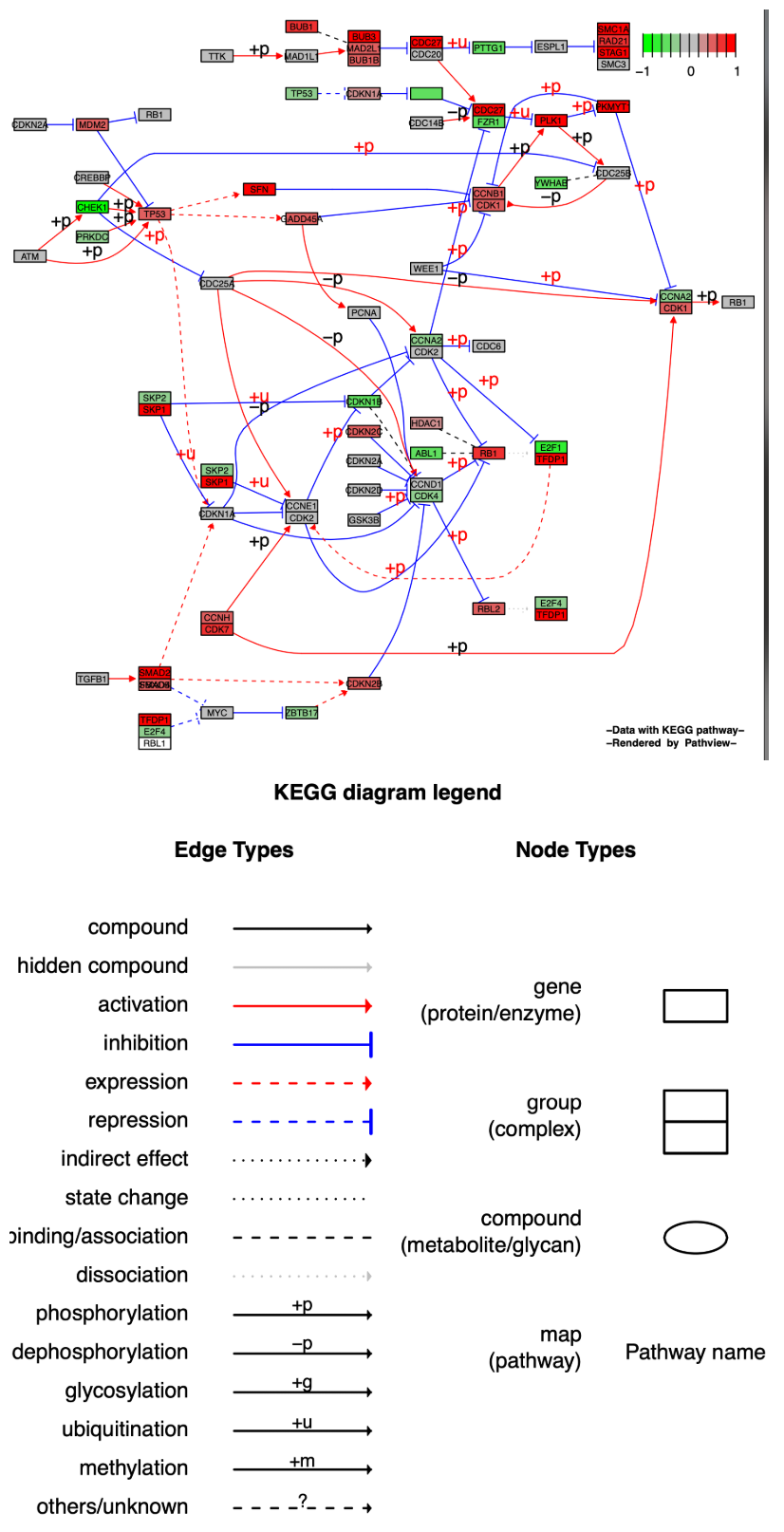
2. 图形调整
考虑到许多改变图形样式的参数仅在kegg.native = FALSE时生效,接下来我们多数情况也基于此来进行绘图
2.1 节点样式调整
p4 <- pathview(gene.data = gse16873.d[, 1],
pathway.id = "04110",
species = "hsa",
out.suffix = "gse16873_node",
kegg.native = F ,
same.layer = F ,
low = '#4575B4', #
mid = '#F7F7BD', #
high = '#D73027', #
cex = 0.5 ,# 字体大小
afactor = 1.6 ,# 节点大小
limit = list( gene = 2 ) # 图例上下限
)
p4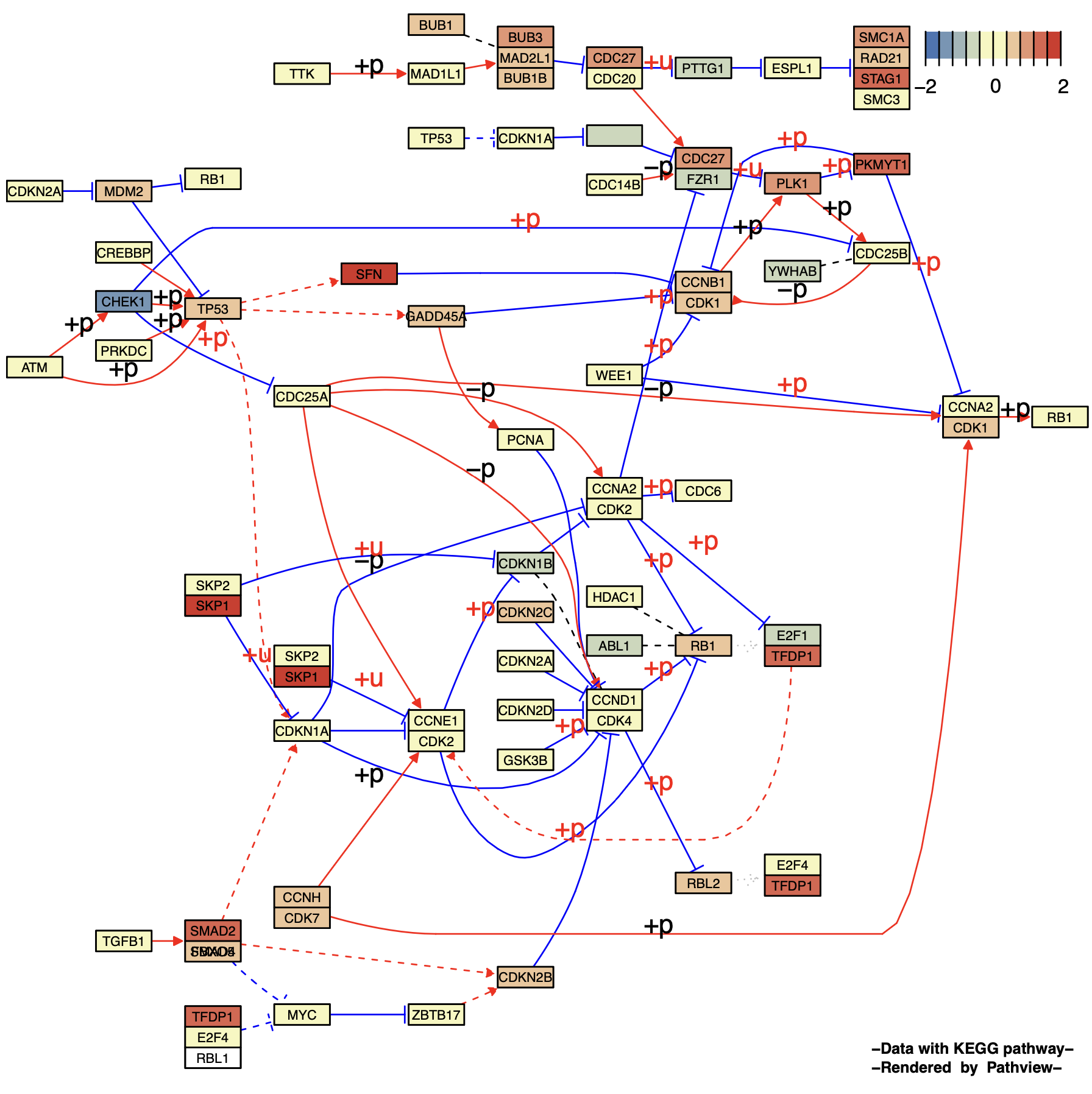
2.2 节点的拆分与扩展
通过设置参数split.group来实现控制节点组拆分为独立的节点,设置expand.node可以将多基因节点扩展为单个基因。
节点组的拆分 (split.group = TRUE) :
p5 <- pathview(gene.data = gse16873.d[, 1],
pathway.id = "04110",
species = "hsa",
out.suffix = "gse16873_split",
kegg.native = F ,
same.layer = F ,
split.group = T # TRUE
)
p5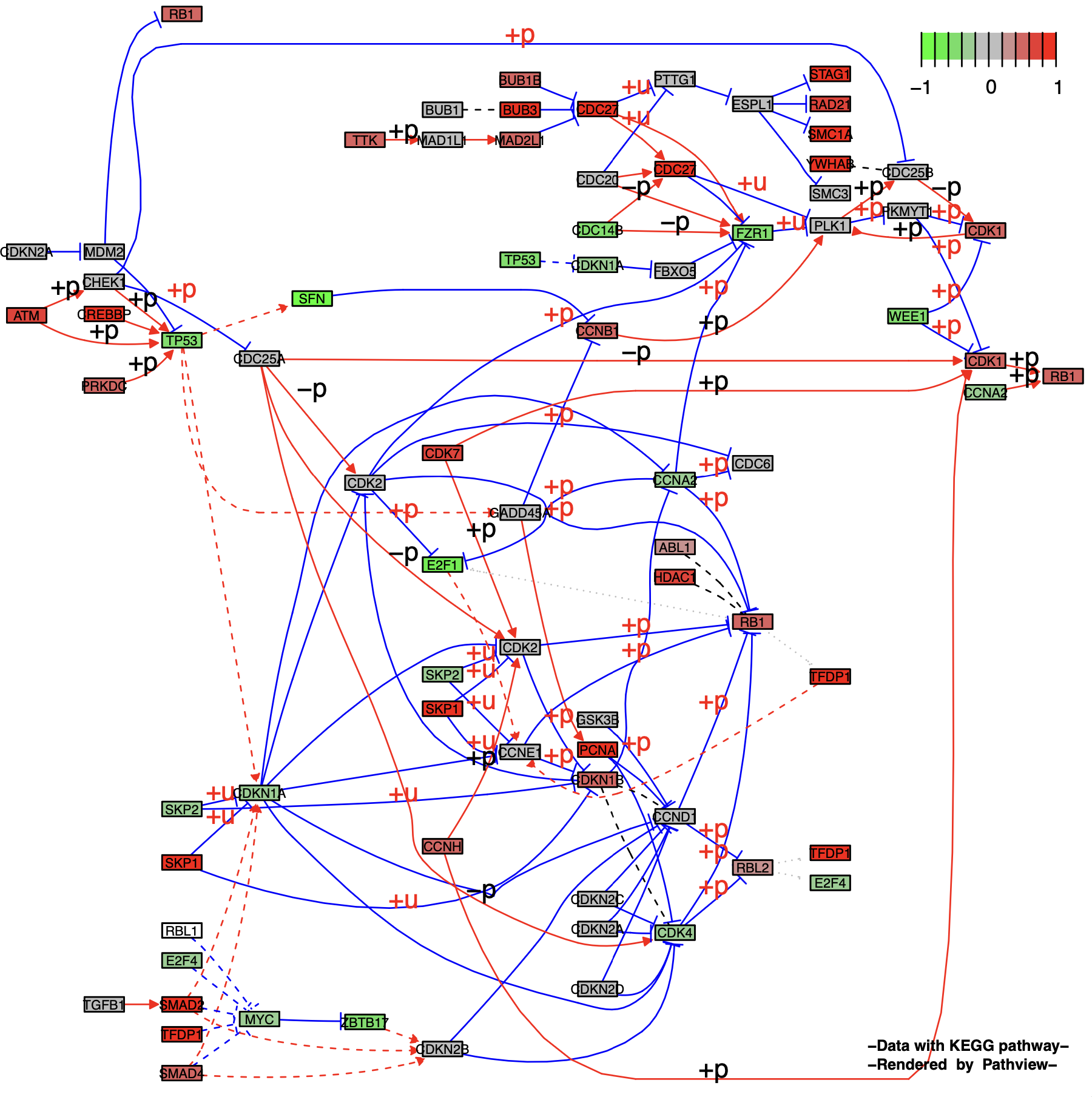
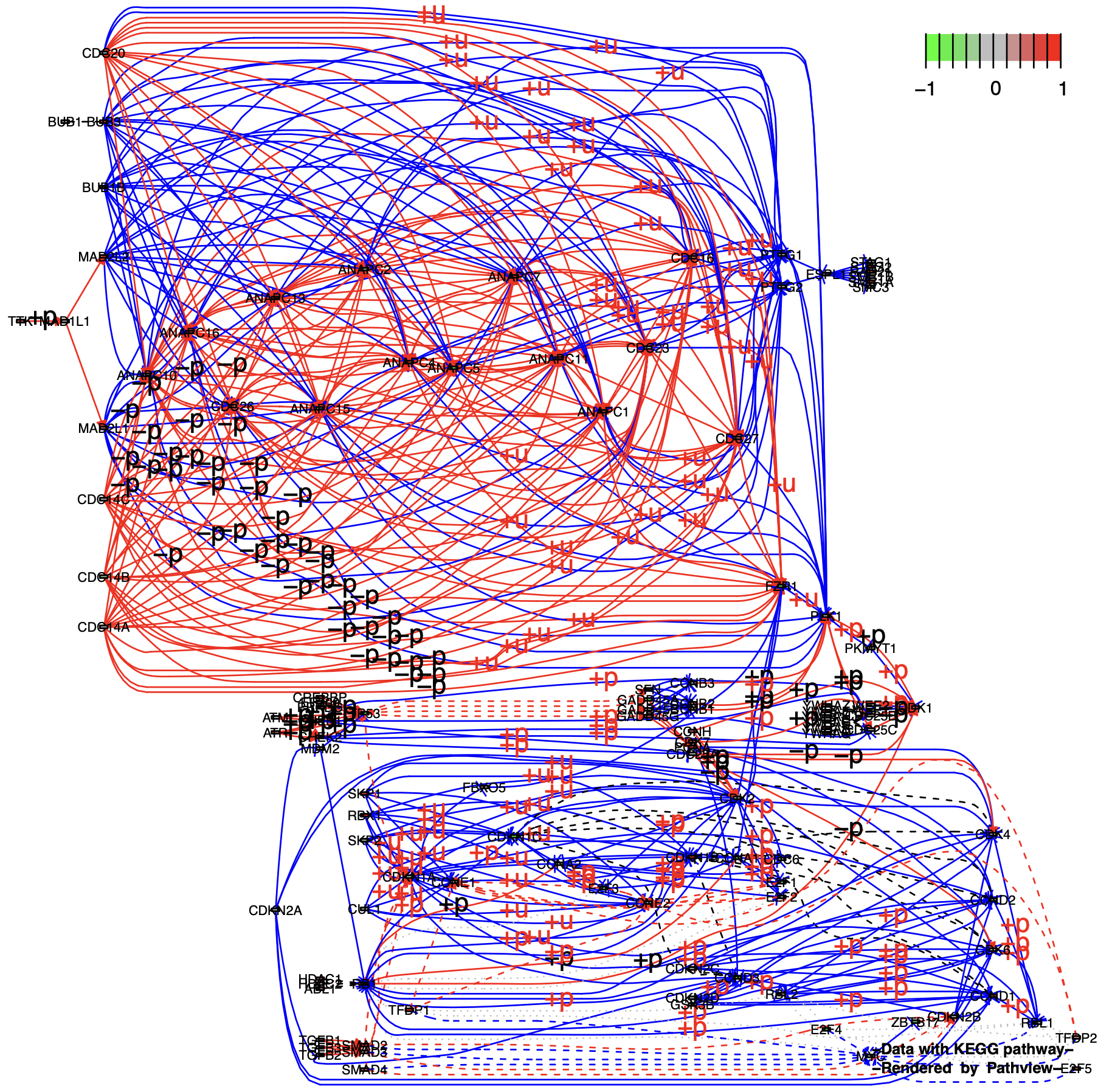
多基因节点扩展为单个基因:(split.group = TRUE & expand.node = TRUE):
p6 <- pathview(gene.data = gse16873.d[, 1],
pathway.id = "04110",
species = "hsa",
out.suffix = "gse16873_expand",
kegg.native = F ,
same.layer = F ,
split.group = T,
expand.node = T # TRUE
)
unique(p6$plot.data.gene$kegg.names) %>% length()
# [1] 88
unique(p7$plot.data.gene$kegg.names) %>% length()
# [1] 158
# 多出的节点即为原多基因节点的扩展
setdiff(unique(B),intersect(A,B))然后发现线条变得错综复杂,以至于我们重点关注的节点和节点之间的关系被淹没了。
2.3 多组样本
以上例子仅绘制了单个样本的通路图,如果是为了比较两个或多个样本的通路变化情况,则需要设置multi.state = TRUE。可以将多个样本的数据整合到同一张通路图中。此时每个节点会被分割成多个片段,每个片段对应一个样本。
p8 <- pathview(gene.data = gse16873.d[, 1:3], # 多样本
pathway.id = "04110",
species = "hsa",
out.suffix = "gse16873_samples",
kegg.native = F ,
same.layer = F ,
multi.state = T #
)呈现的效果是:节点被分割成多个对应样本数量的片段,一个样本对应一个色块
2.4 化合物的通路图
化合物数据
之前我们只介绍了基因数据的通路可视化,如果单纯做化合物数据的话,其实是跟基因一样的,只需要修改gene.data为cpd.data。
# 模拟6个样本的化合物数据
data("cpd.simtypes")
cpd_data <- sim.mol.data(mol.type="cpd", nmol=3000)
cpd_data2 <- matrix(sample(cpd_data,18000,replace = T), ncol = 6)
rownames(cpd_data2) = names(cpd_data)
colnames(cpd_data2) = colnames(gene_data)
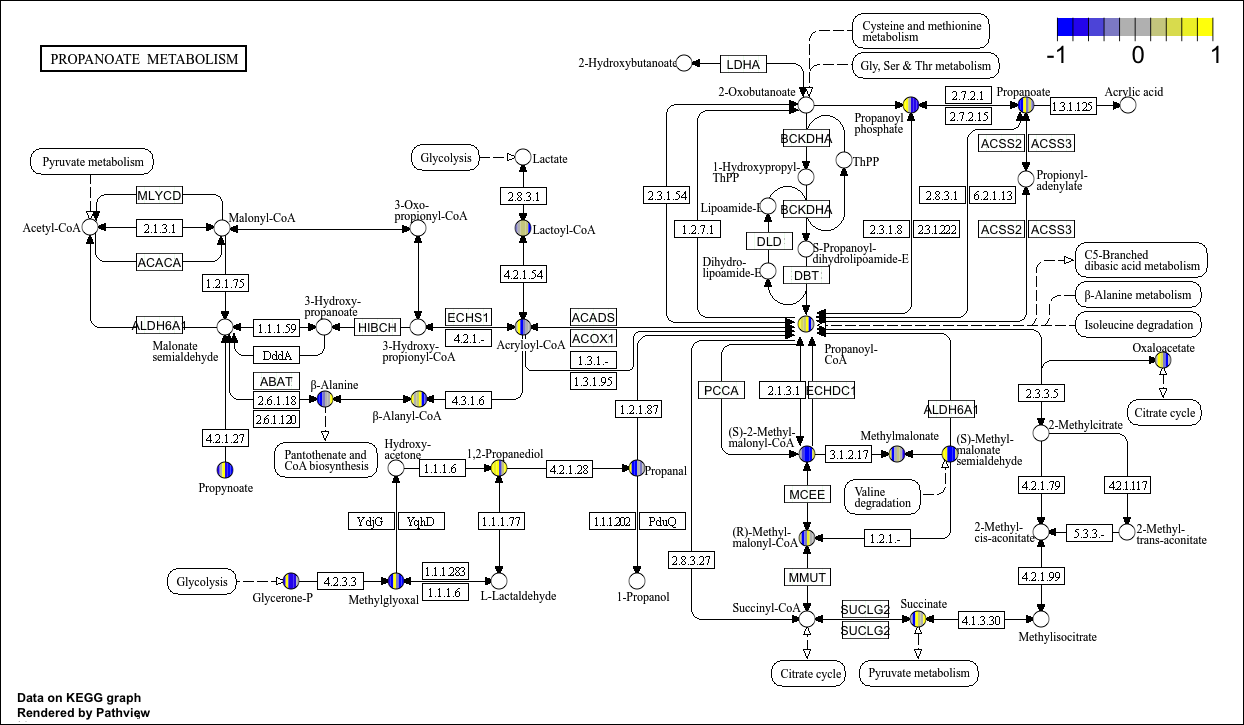
head(cpd_data2)KEGG视图
p9 <- pathview(cpd.data = cpd_data2, # 化合物数据
pathway.id = "00640", #
species = "hsa",
out.suffix = "gse16873_KEGG_cpd",
kegg.native = T , # KEGG视图
same.layer = F
) 
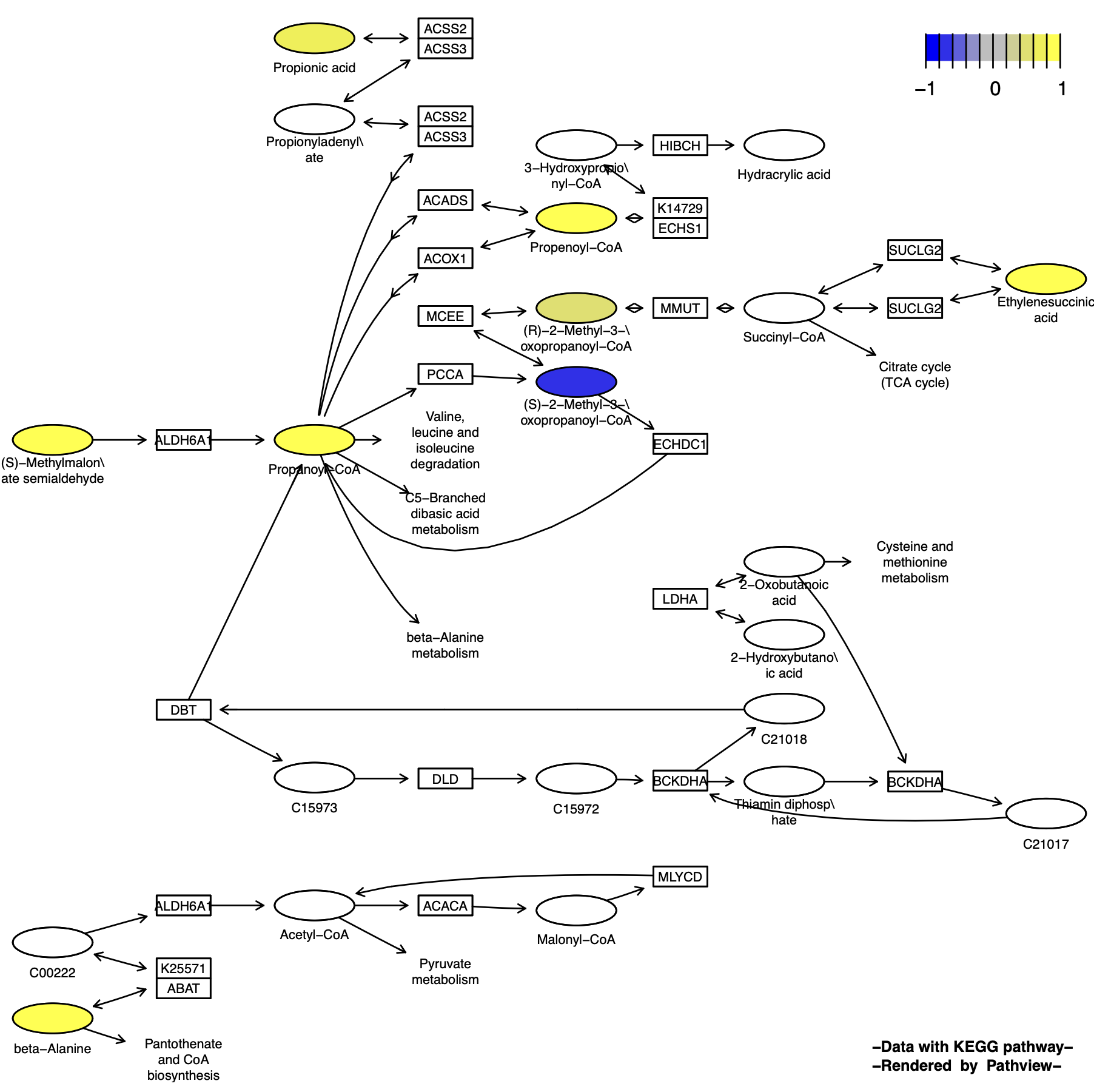
Graphviz视图
p10 <- pathview(cpd.data = cpd_data, # 化合物数据
pathway.id = "00640",
species = "hsa",
out.suffix = "gse16873_Graphviz_cpd",
kegg.native = F , # Graphviz视图
same.layer = F ,
cpd.lab.offset = -1.0 #化合物标签位置
) 
化合物和基因数据
接下来我们举例说明同时绘制基因和化合物数据的情况:
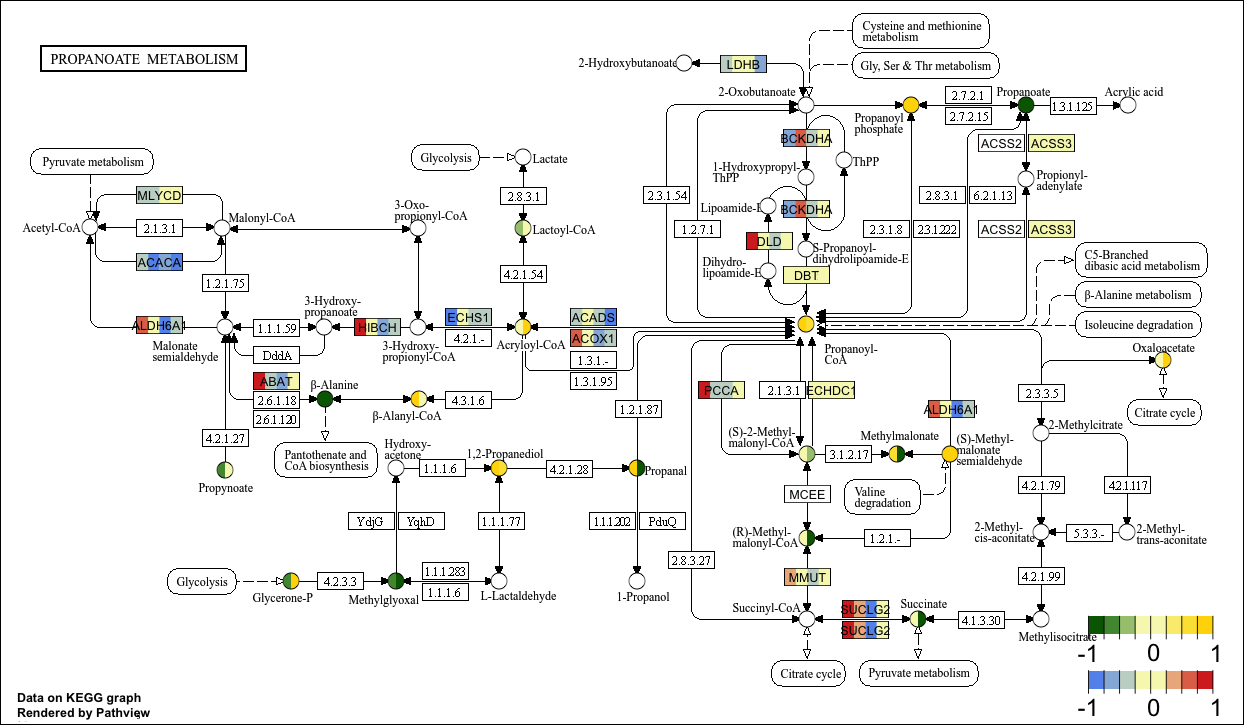
KEGG视图
p11 <- pathview(gene.data = gene_data[,1:4], # 基因数据
cpd.data = cpd_data2[,1:2], # 化合物数据
pathway.id = "00640",
species = "hsa",
out.suffix = "gse16873_KEGG_gene_cpd",
kegg.native = T , # KEGG视图
same.layer = F ,
cpd.lab.offset = -1.0 ,
key.pos = 'bottomright',
match.data = F, # 不匹配样本数
multi.state = T ,
bins = list(gene = 8, cpd = 8), # 改变图例色块的数量
low = list(gene = "#6495ED", cpd = "#006400"),
mid = list(gene = "#F7F7BD", cpd = "#F7F7BD"),
high = list(gene = "#D73027", cpd ="#FFD700")
) 
Graphviz视图
p12 <- pathview(gene.data = gene_data[,1:4], # 基因数据
cpd.data = cpd_data2[,1:2], # 化合物数据
pathway.id = "00640",
species = "hsa",
out.suffix = "gse16873_Graphviz_gene_cpd",
kegg.native = F , # Graphviz视图
same.layer = F ,
cpd.lab.offset = -1.0 ,
key.pos = 'topright',
match.data = F, # 不匹配样本数
multi.state = T ,
bins = list(gene = 10, cpd = 10), # 改变图例色块的数量
low = list(gene = "#6495ED", cpd = "#006400"),
mid = list(gene = "#F7F7BD", cpd = "#F7F7BD"),
high = list(gene = "#D73027", cpd ="#FFD700")
) 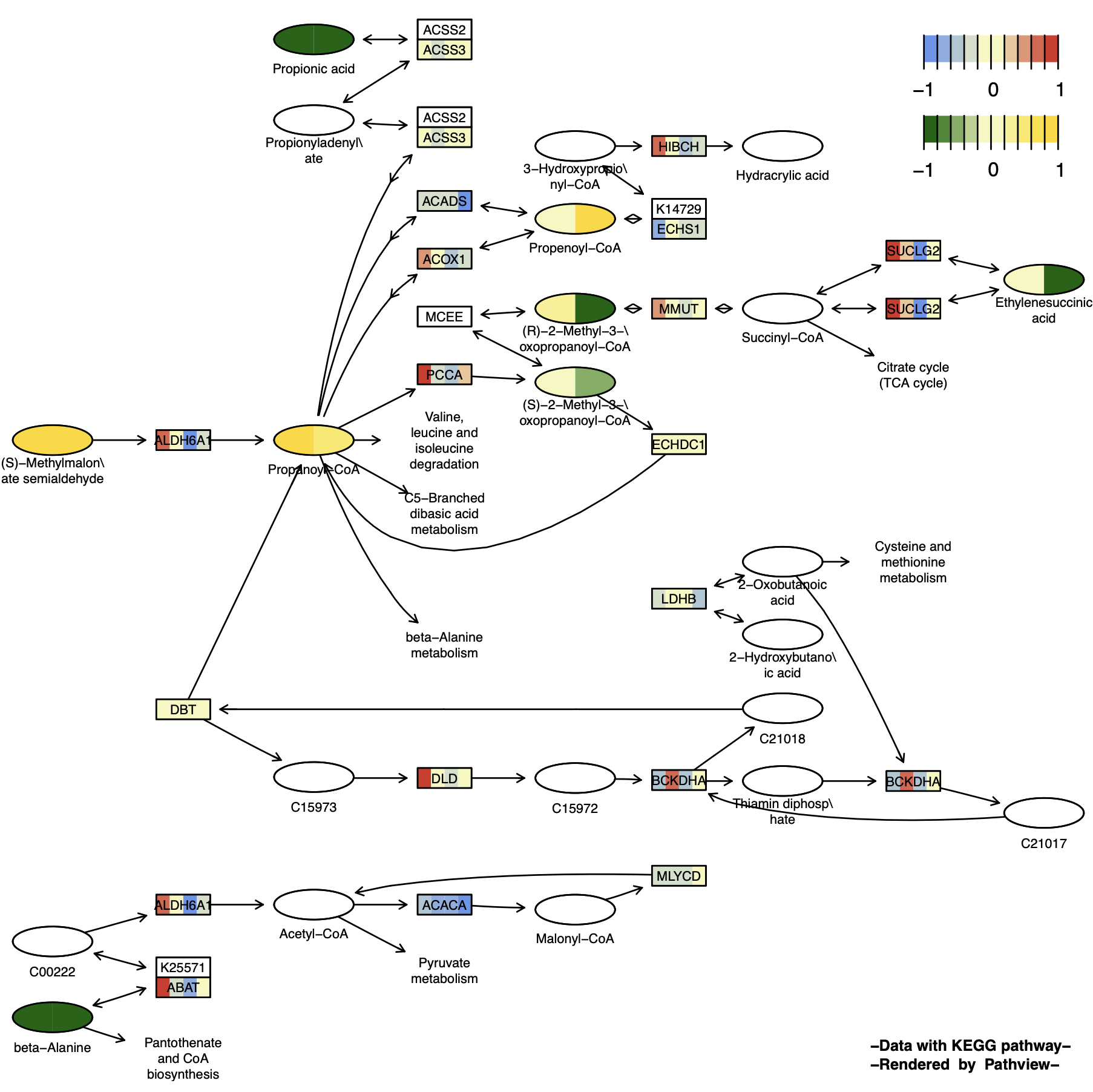
当gene和cpd数据的样本数不一致时,此时参数应设置为match.data=FALSE,若设置为match.data = TRUE,则样本数少的数据会生成相差的NA列以配对样本数量。默认NA数据为透明无色,可通过na.col设置颜色。
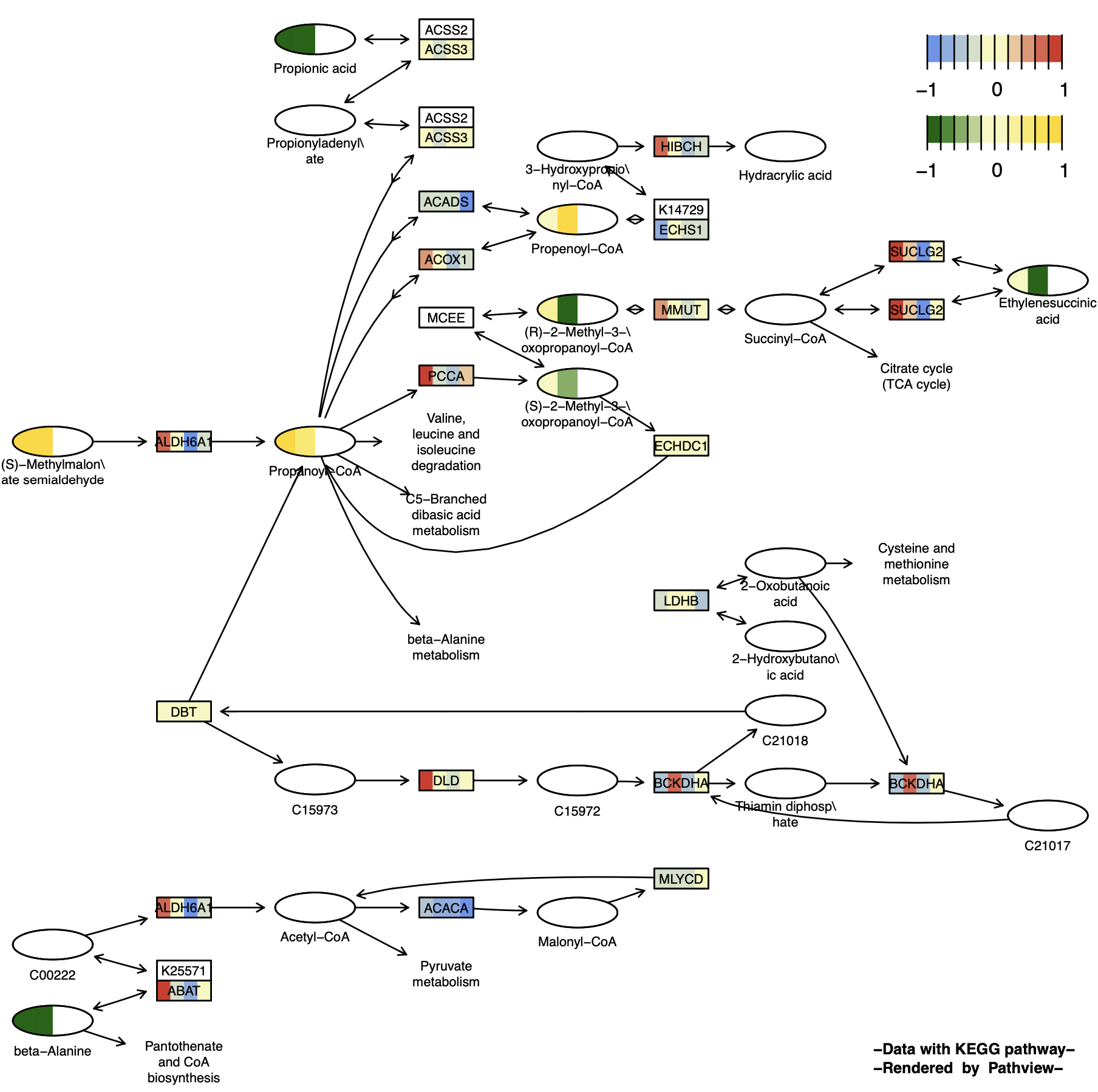
p13 <- pathview(gene.data = gene_data[,1:4],
cpd.data = cpd_data2[,1:2],
pathway.id = "00640",
species = "hsa",
out.suffix = "gse16873_Graphviz_gene_cpd_match",
kegg.native = F , # Graphviz视图
same.layer = F ,
cpd.lab.offset = -1.0 ,
key.pos = 'topright',
match.data = T, # 匹配样本数:为cpd添加2列NA
multi.state = T ,
bins = list(gene = 10, cpd = 10),
na.col = 'transparent' , # NA数据颜色设置
low = list(gene = "#6495ED", cpd = "#006400"),
mid = list(gene = "#F7F7BD", cpd = "#F7F7BD"),
high = list(gene = "#D73027", cpd ="#FFD700")
) 可以发现化合物节点只有一半是有颜色的,因为另一半是NA,默认为透明无色。
2.5 离散数据
如果我们的输入数据不是像上述例子的连续变量,而是如high/low的分类变量(可用1/0替换)或显著差异的基因名称,则需要设置discrete参数以绘制离散变量的通路视图。
这里我们取基因数据为离散变量1/0,而化合物为连续变量。
# 获取基因数据 (离散变量)
data("gse16873.d")
gene_data <- gse16873.d
colnames(gene_data)
gene_data2 <- apply(gene_data, 2, function(x){
ifelse(x > mean(x), 1 , 0 )
}) %>% as.data.frame()
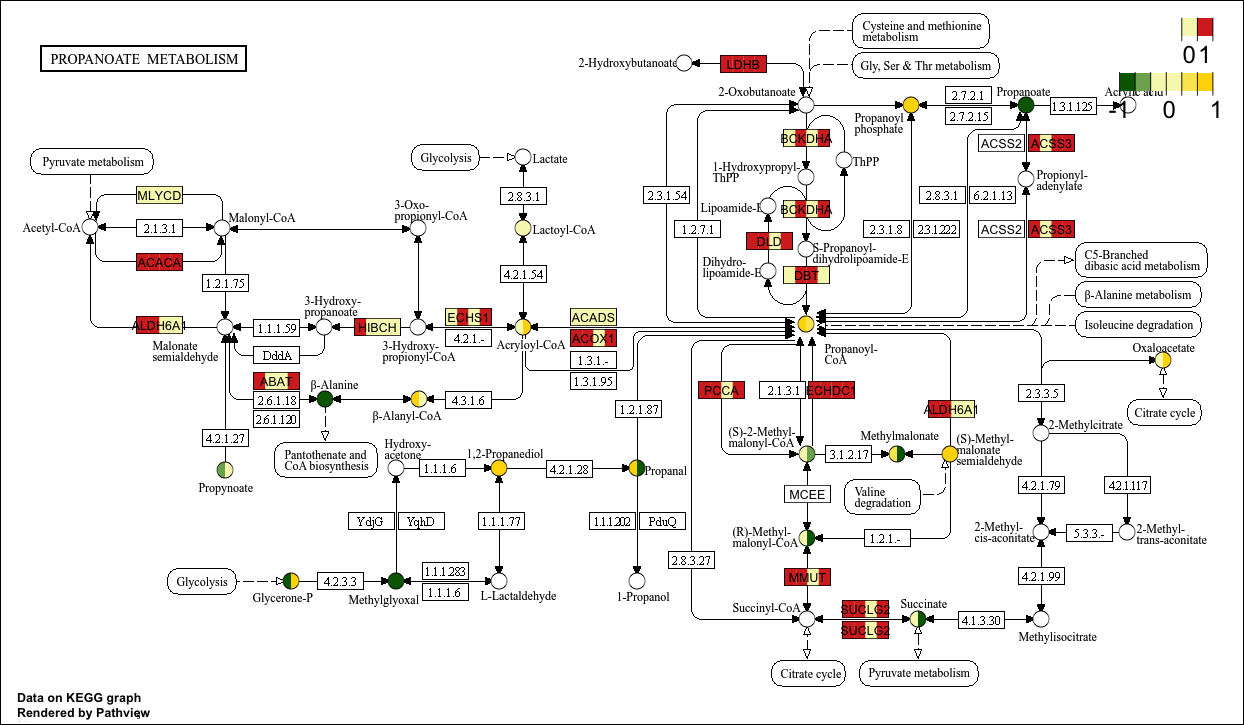
str(gene_data2)p14 <- pathview(gene.data = gene_data2[,1:4], # 离散变量
cpd.data = cpd_data2[,1:2], # 连续变量
pathway.id = "00640",
species = "hsa",
out.suffix = "gse16873_KEGG_gene_cpd_discrete",
kegg.native = T , # KEGG视图
same.layer = F ,
cpd.lab.offset = -1.0 ,
key.pos = 'topright',
match.data = F,
multi.state = T ,
bins = list(gene = 1, cpd = 6),
low = list(gene = "#F7F7BD", cpd = "#006400"),
mid = list(gene = "#F7F7BD", cpd = "#F7F7BD"),
high = list(gene = "#D73027", cpd ="#FFD700"),
discrete = list(gene = T, cpd = F) ,# 离散数据为T
limit = list(gene = 1, cpd = c(-1,1)) ,
both.dirs = list(gene = F, cpd = T) # gene为单向数据
) 
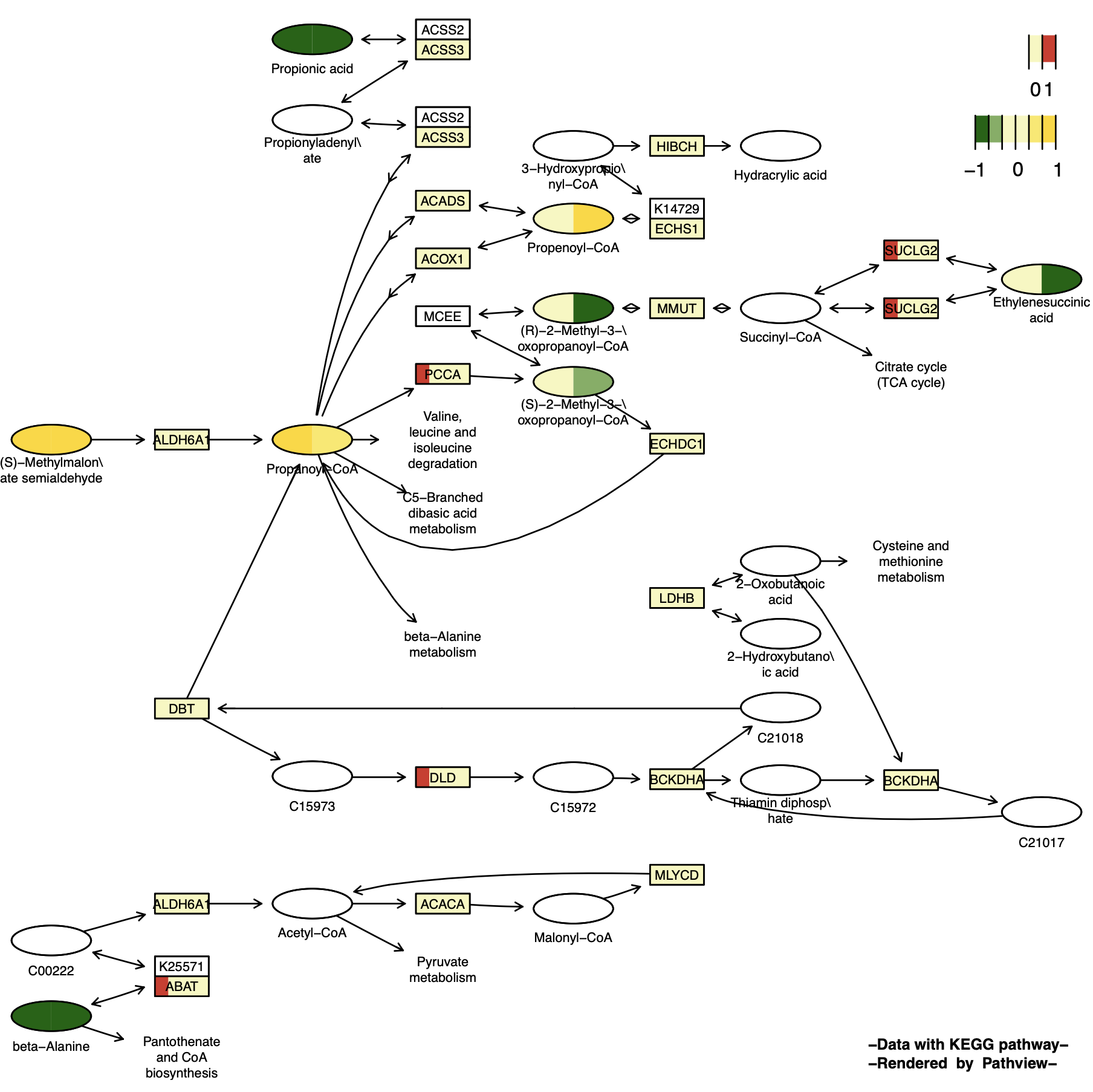
p15 <- pathview(gene.data = gene_data[,1:4], # 离散变量
cpd.data = cpd_data2[,1:2], # 连续变量
pathway.id = "00640",
species = "hsa",
out.suffix = "gse16873_Graphviz_gene_cpd_discrete",
kegg.native = F , # Graphviz视图
same.layer = F ,
cpd.lab.offset = -1.0 ,
key.pos = 'topright',
match.data = F,
multi.state = T ,
bins = list(gene = 1, cpd = 6),
low = list(gene = "#F7F7BD", cpd = "#006400"),
mid = list(gene = "#F7F7BD", cpd = "#F7F7BD"),
high = list(gene = "#D73027", cpd ="#FFD700"),
discrete = list(gene = T, cpd = F) ,# 离散数据为T
limit = list(gene = 1, cpd = c(-1,1)),
both.dirs = list(gene = F, cpd = T) # gene.data为单向数据
) 
应用场景
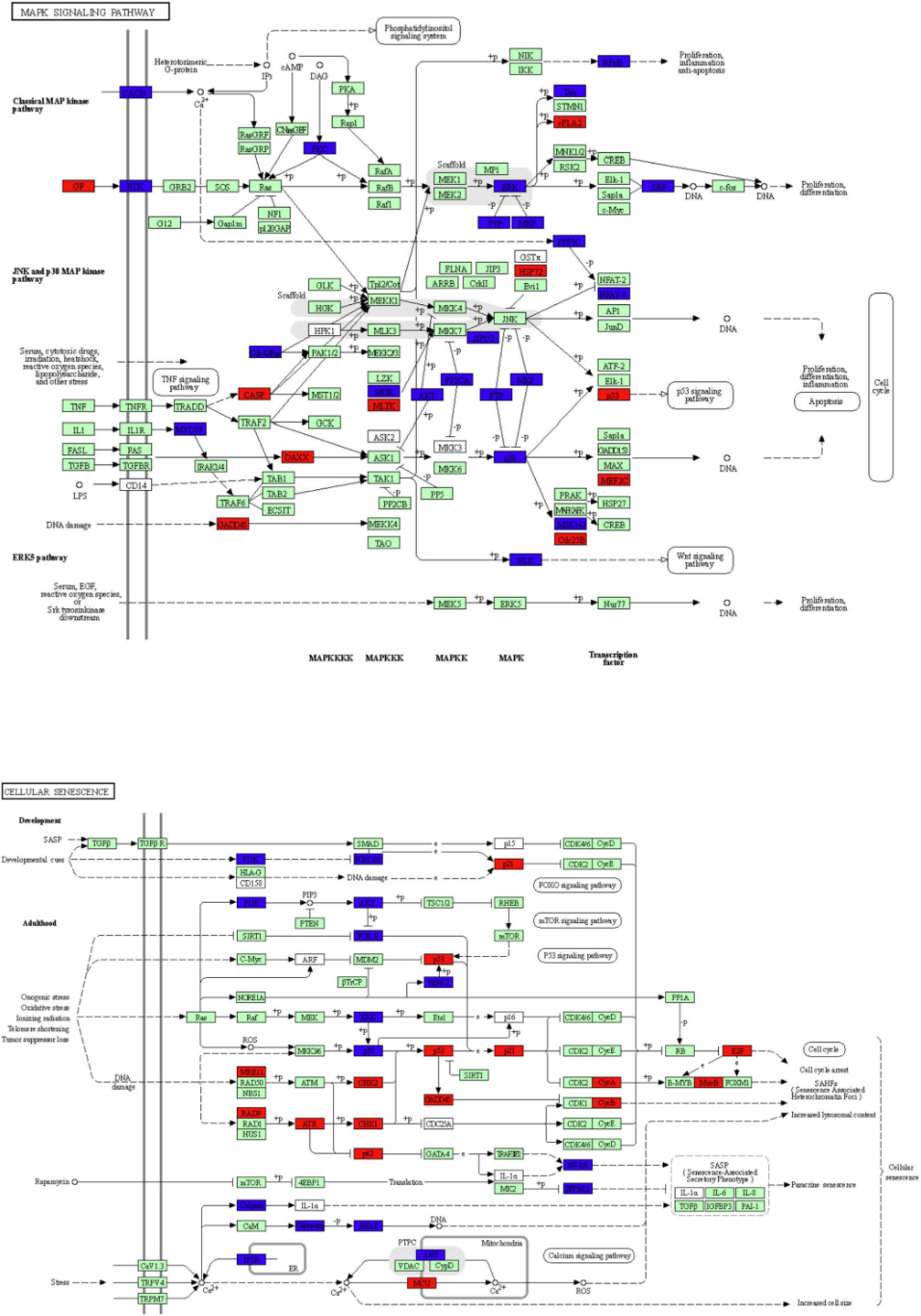
该图展示了关键的KEGG途径:MAPK信号通路和细胞衰老信号通路。红色节点代表cingulin b-mo突变体中上调的DEG,绿色标记的节点代表cingulin b-mo突变体中下调的DEG,而蓝色标记的节点代表对照MO和c链蛋白B-MO胚胎之间的重叠靶标。该分析是从不同组的三个独立实验进行的,每个组都有30个胚胎。[2]
参考文献
[1] Luo, W., & Brouwer, C. (2013). Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics (Oxford, England), 29(14), 1830–1831. https://doi.org/10.1093/bioinformatics/btt285IF: 4.4 B3.
[2] Lu Y, Tang D, Zheng Z, et al. Cingulin b Is Required for Zebrafish Lateral Line Development Through Regulation of Mitogen-Activated Protein Kinase and Cellular Senescence Signaling Pathways. Front Mol Neurosci. 2022;15:844668. Published 2022 May 5. doi:10.3389/fnmol.2022.844668.