# 安装包
if (!requireNamespace("umap", quietly = TRUE)) {
install.packages("umap")
}
if (!requireNamespace("ggplot2", quietly = TRUE)) {
install.packages("ggplot2")
}
if (!requireNamespace("patchwork", quietly = TRUE)) {
install.packages("patchwork")
}
if (!requireNamespace("RColorBrewer", quietly = TRUE)) {
install.packages("RColorBrewer")
}
if (!requireNamespace("Seurat", quietly = TRUE)) {
install.packages("Seurat")
}
if (!requireNamespace("SeuratData", quietly = TRUE)) {
remotes::install_github('satijalab/seurat-data')
}
if (!requireNamespace("dplyr", quietly = TRUE)) {
install.packages("dplyr")
}
if (!requireNamespace("mlbench", quietly = TRUE)) {
install.packages("mlbench")
}
# 加载包
library(umap)
library(ggplot2)
library(patchwork)
library(RColorBrewer)
library(Seurat)
library(SeuratData)
library(dplyr)
library(mlbench)UMAP图
示例
UMAP(Uniform Manifold Approximation and Projection,均匀流形近似和投影)是一种强大的非线性降维技术,主要应用于处理高维数据。UMAP算法的核心在于保持数据的局部和全局结构,通过构建数据点之间的邻近关系图,利用图的拓扑结构进行流形近似和优化。它能够将高维数据有效地映射到低维空间,以便于可视化和进一步分析,在生物信息学中,非常适合用于处理基因表达、微生物组等具有高维特征的数据。下面我们将分别展示在二分类的临床数据和单细胞测序数据中UMAP的应用。
环境配置
系统要求: 跨平台(Linux/MacOS/Windows)
编程语言:R
依赖包:
umap,ggplot2,patchwork,RColorBrewer,Seurat,SeuratData,dplyr,mlbench
sessioninfo::session_info("attached")─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.6.0 (2026-04-24)
os Ubuntu 24.04.4 LTS
system x86_64, linux-gnu
ui X11
language (EN)
collate C.UTF-8
ctype C.UTF-8
tz UTC
date 2026-05-09
pandoc 3.1.3 @ /usr/bin/ (via rmarkdown)
quarto 1.9.37 @ /usr/local/bin/quarto
──────────────────────────────────────────────────────────────────────────────数据准备
1. 临床表型数据:威斯康星乳腺癌数据集
使用mlbench包中的BreastCancer数据集。该数据最初由威斯康星州医院的Dr. William H. Wolberg收集,包含了从乳腺癌患者收集的肿瘤特征的测量值,如肿瘤的半径、纹理、对称性等,以及相应的良性(benign)或恶性(malignant)标签。为了便于分析,下面对数据进行简单的预处理:
data(BreastCancer)
wdbc_data <- BreastCancer[, -1] # 移除ID列
wdbc_data <- na.omit(wdbc_data)
features <- wdbc_data[, 1:9] # 使用前9个特征
features <- as.data.frame(lapply(features, function(x) as.numeric(as.character(x))))
diagnosis <- wdbc_data$Class
head(features) 2. 单细胞测序数据:IFNB数据集
IFNB是基于PBMC的数据集,PBMC(Peripheral Blood Mononuclear Cells)是由10x Genomics提供的外周血单个核细胞scRNA-seq数据集,包含了已注释的B 细胞、T 细胞、NK 细胞、单核细胞等。IFNB包含了两个PBMC数据,分别来自干扰素刺激组和对照组。
单细胞测序数据由于其高维、稀疏的特点,非常适合应用于UMAP降维技术。下面将以IFNB单细胞RNA-seq数据集为例展示单细胞分析中的UMAP降维。该数据集可以通过SeuratData加载。
RunUMAP()是Seurat提供的UMAP降维函数。下面的代码中提供了单细胞数据UMAP前预处理的简单说明,具体原理及说明可以参照专业的单细胞数据分析教程,这里不再赘述。
# InstallData("ifnb")
# ifnb <- LoadData("ifnb") # Seurat V4
data("ifnb")
ifnb <- UpdateSeuratObject(ifnb) # Seurat V5
ifnb <- NormalizeData(ifnb) # 对数据进行Log2(x+1)变换
ifnb <- FindVariableFeatures(ifnb) # 提取方差最大的特征用于降维
ifnb <- ScaleData(ifnb) # 数据缩放
ifnb <- RunPCA(ifnb) # PCA预先降维
ifnb <- RunUMAP(ifnb, dims = 1:20) # UMAP降维可视化
1. 临床表型数据UMAP可视化
umap是一个用于进行UMAP降维的R包,其中提供了便捷的umap()函数进行降维和调参。
set.seed(123)
wdbc_umap <- umap(features,
n_neighbors = 15,
min_dist = 0.2,
metric = "euclidean")
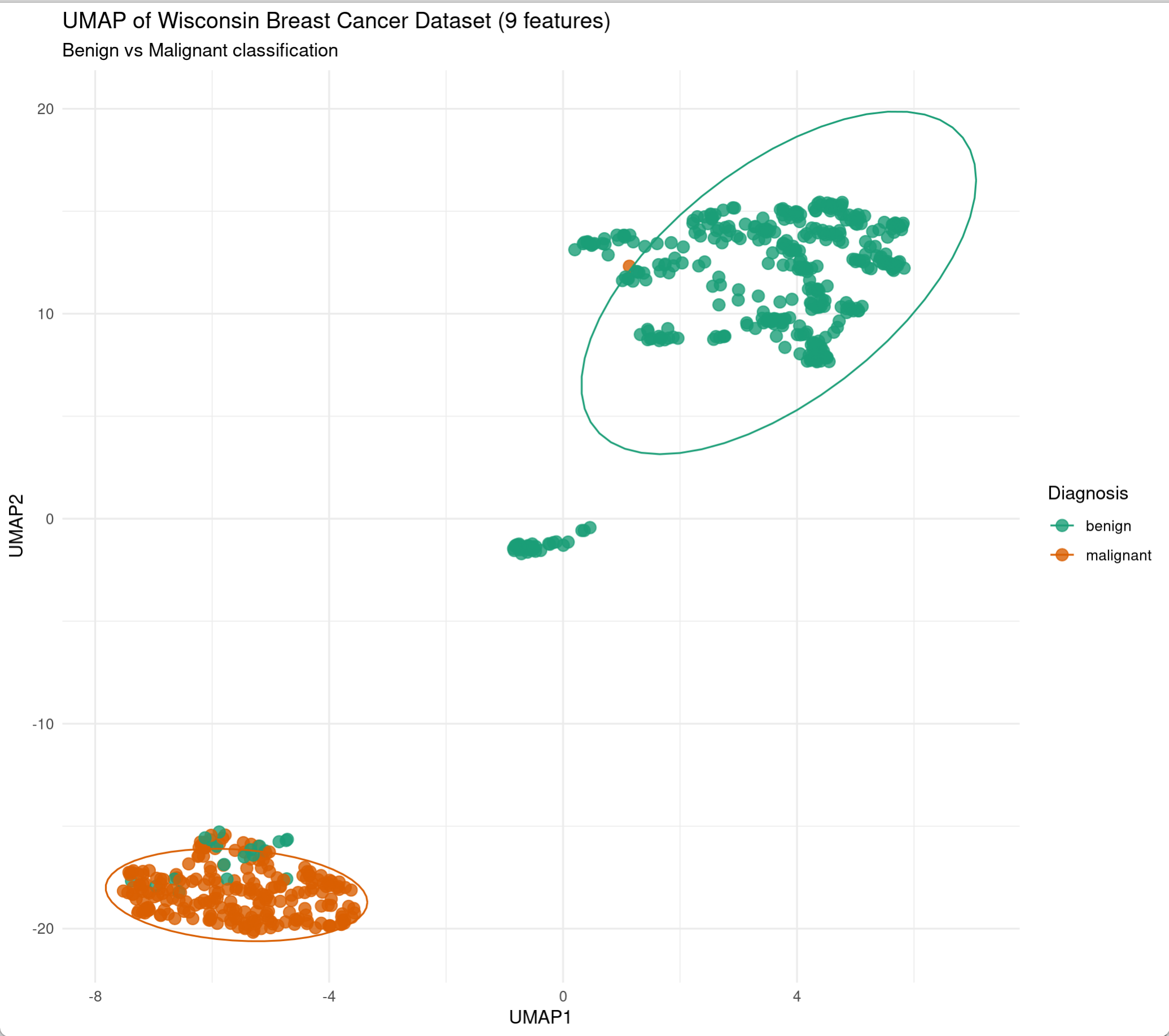
ggplot(data.frame(wdbc_umap$layout, Diagnosis = diagnosis),
aes(X1, X2, color = Diagnosis)) +
geom_point(size = 3, alpha = 0.8) +
stat_ellipse(level = 0.9) +
theme_minimal() +
labs(title = "UMAP of Wisconsin Breast Cancer Dataset",
x = "UMAP1", y = "UMAP2",
subtitle = "n_neighbors=15, min_dist=0.2") +
scale_color_manual(values = c("benign" = "#1b9e77", "malignant" = "#d95f02"))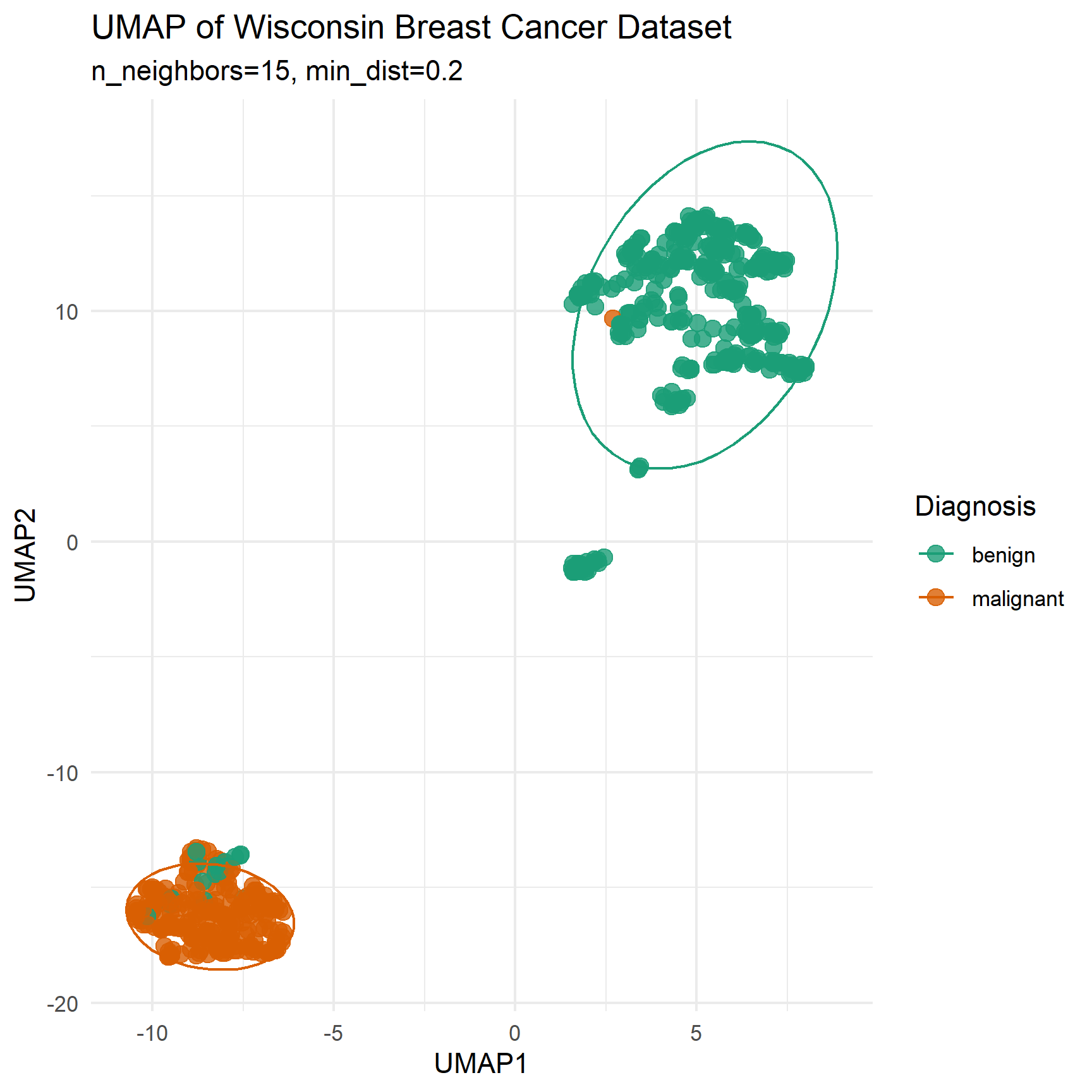
提示
参数说明:
n_neighbors: 用于控制局部结构的粒度,较小值能够捕捉更精细的局部结构,但可能过度拟合噪声;较大值有利于保留更多全局结构,但可能模糊局部细节。默认为15min_dist: 控制流形空间中嵌入点的最小间距,较小值下数据点更紧密,局部结构更清晰;较大值下的点会更分散,全局结构更明显metric: 定义距离度量方式,可选"euclidean","cosine","manhattan","pearson"
参数敏感性分析
我们可以尝试调整UMAP中的不同参数来调整降维效果,下面展示了采用三种不同的参数组合得到的UMAP结果比较。
# 定义参数组合
params <- list(
list(n_neighbors=5, min_dist=0.1, label="Aggressive"),
list(n_neighbors=15, min_dist=0.2, label="Default"),
list(n_neighbors=50, min_dist=0.5, label="Conservative")
)
# 生成UMAP并绘图
plots <- list()
for (i in seq_along(params)) {
set.seed(123)
umap_res <- umap::umap(features,
n_neighbors = params[[i]]$n_neighbors,
min_dist = params[[i]]$min_dist)
df <- data.frame(umap_res$layout, Diagnosis = diagnosis)
plots[[i]] <- ggplot(df, aes(X1, X2, color = Diagnosis)) +
geom_point(size = 2, alpha = 0.7) +
labs(title = paste("Params:", params[[i]]$label),
subtitle = sprintf("n_neighbors=%d, min_dist=%.1f",
params[[i]]$n_neighbors, params[[i]]$min_dist),
x = "UMAP1", y = "UMAP2") +
theme_minimal(base_size = 10) +
scale_color_manual(values = c("benign" = "#1b9e77", "malignant" = "#d95f02"))
}
# 使用patchwork排版
(plots[[1]] / plots[[2]] / plots[[3]])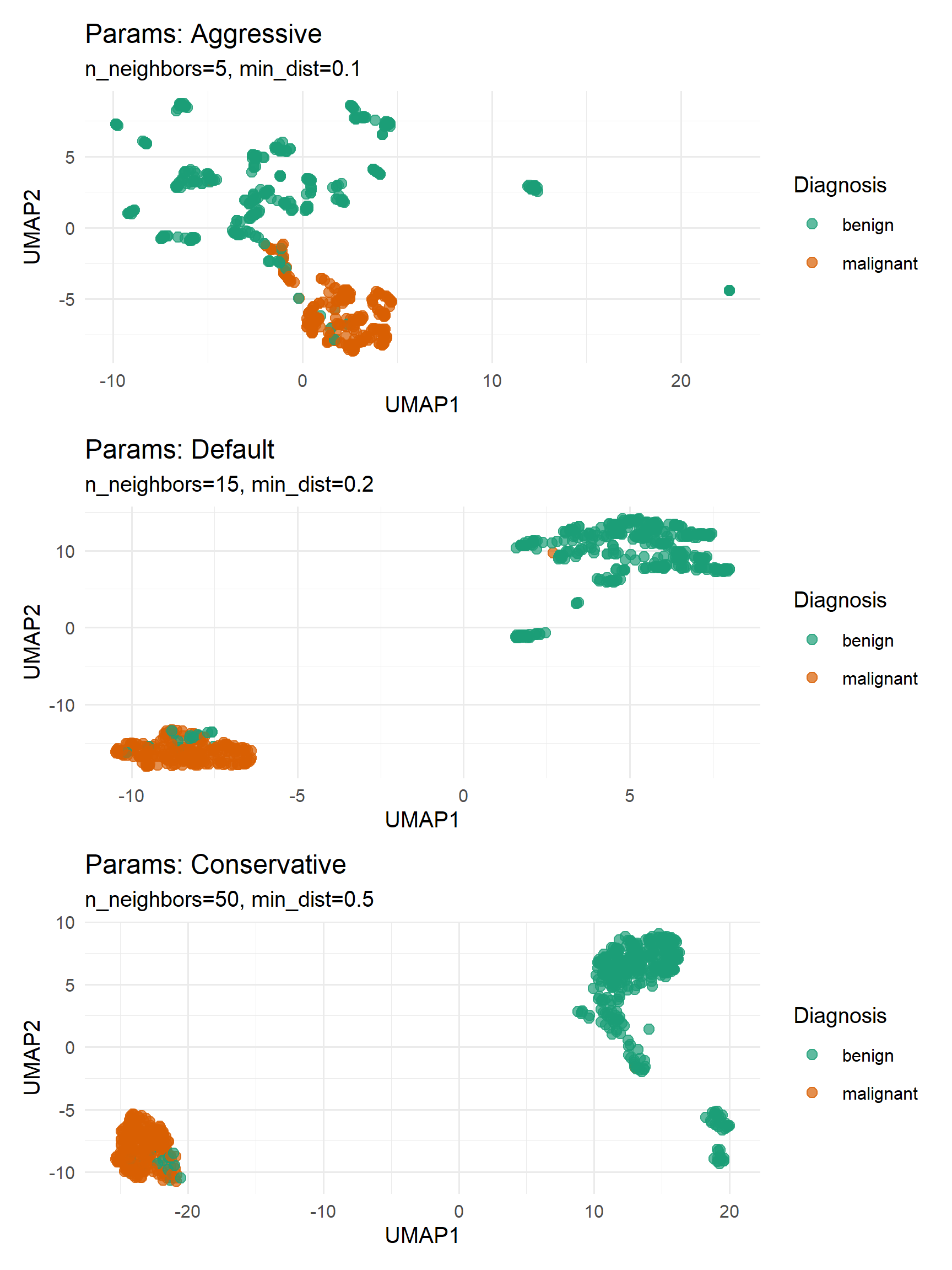
2. Seurat UMAP可视化
Seurat中提供了UMAP降维可视化的接口函数DimPlot(),能帮助用户快速可视化单细胞数据。 下面分别展示了我们上面得到的同一个降维空间中,将细胞分别按照细胞类型和处理组别着色的结果。
(注意:上面的代码中并未进行细胞的聚类,类别结果均来自数据集中预设的注释)
plots <- list()
group_ident <- c('seurat_annotations', 'stim')
for (i in seq_along(group_ident)) {
p <- DimPlot(ifnb, reduction = 'umap', group.by = group_ident[i])
plots[[i]] <- p
}
(plots[[1]] / plots[[2]])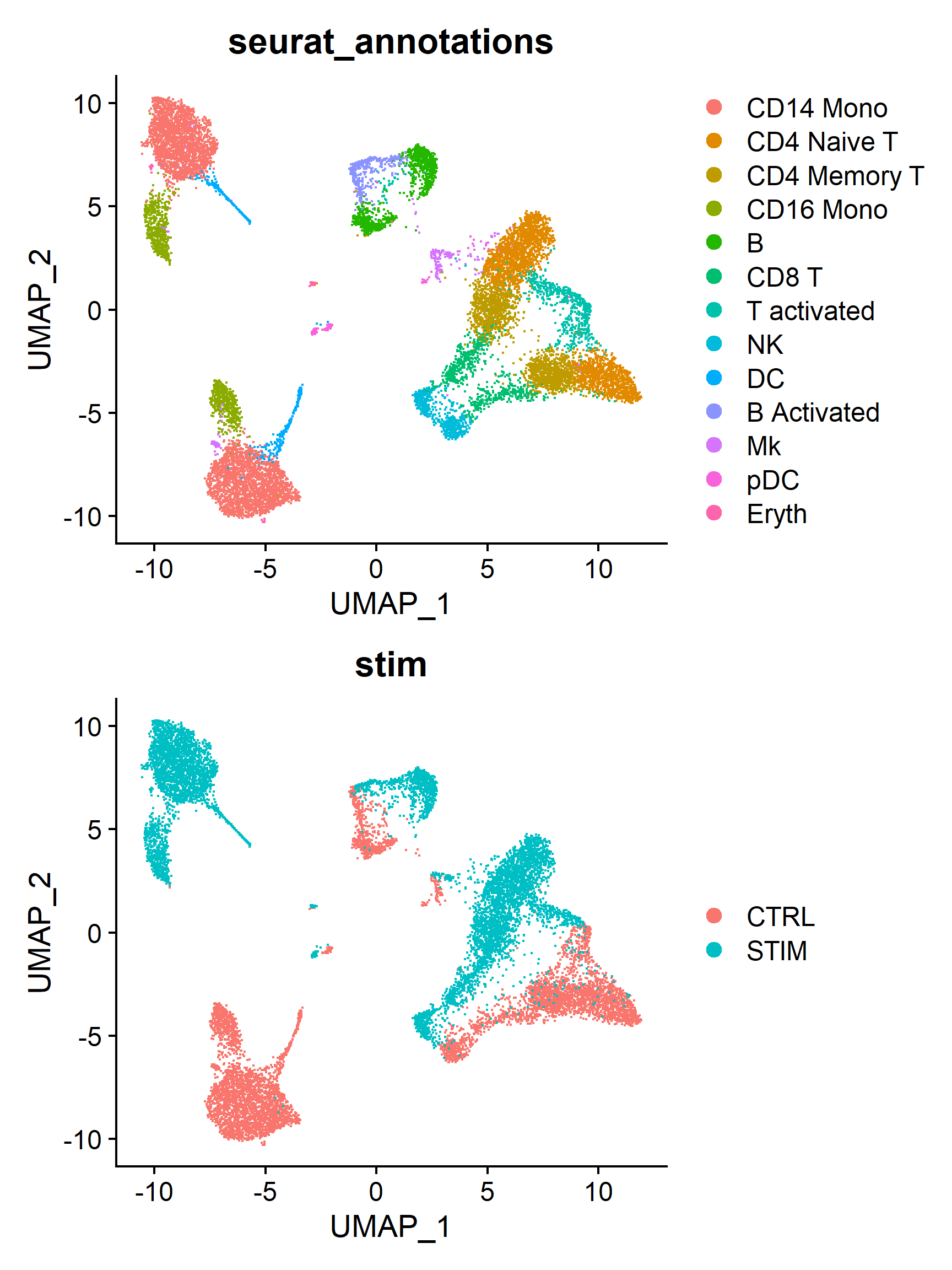
3. ggplot2自定义UMAP可视化
除了利用接口函数,我们还可以使用ggplot2自定义绘制UMAP图。
# 提取UMAP坐标和元数据
umap_df <- Embeddings(ifnb, "umap") %>% as.data.frame() %>% mutate(CellType = ifnb$seurat_annotations, Treatment = ifnb$stim)
head(umap_df)
custom_colors <- c(
"#8DD3C7", "#FFFFB3", "#BEBADA", "#FB8072", "#80B1D3",
"#FDB462", "#B3DE69", "#FCCDE5", "#D9D9D9", "#BC80BD",
"#CCEBC5", "#FFED6F", "#A6CEE3")
ggplot(umap_df, aes(UMAP_1, UMAP_2, color = CellType)) +
geom_point(size = 1.5, alpha = 0.8) +
theme_minimal() + labs(title = "IFNB UMAP", x = "UMAP1", y = "UMAP2") +
scale_color_manual(values = custom_colors)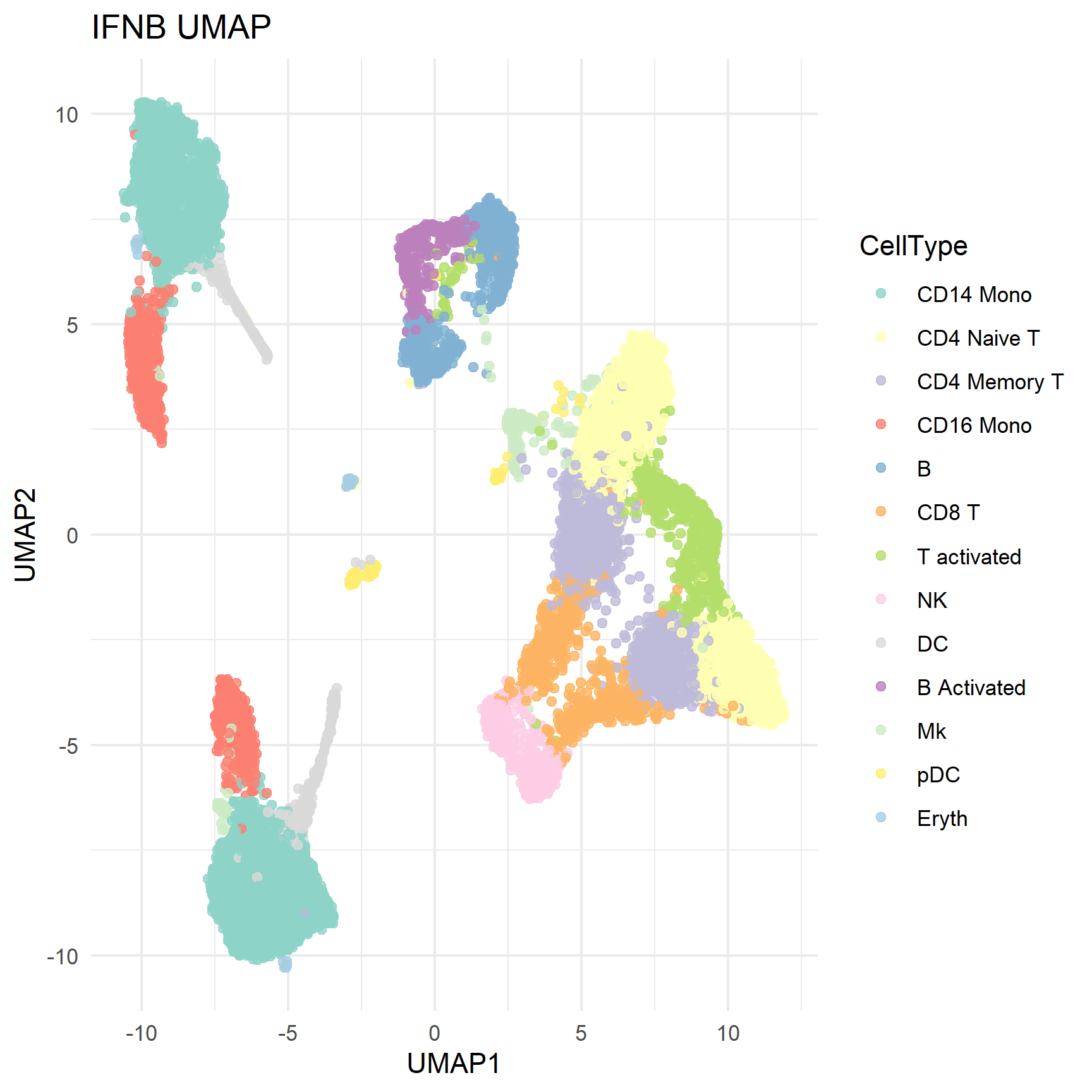
应用场景
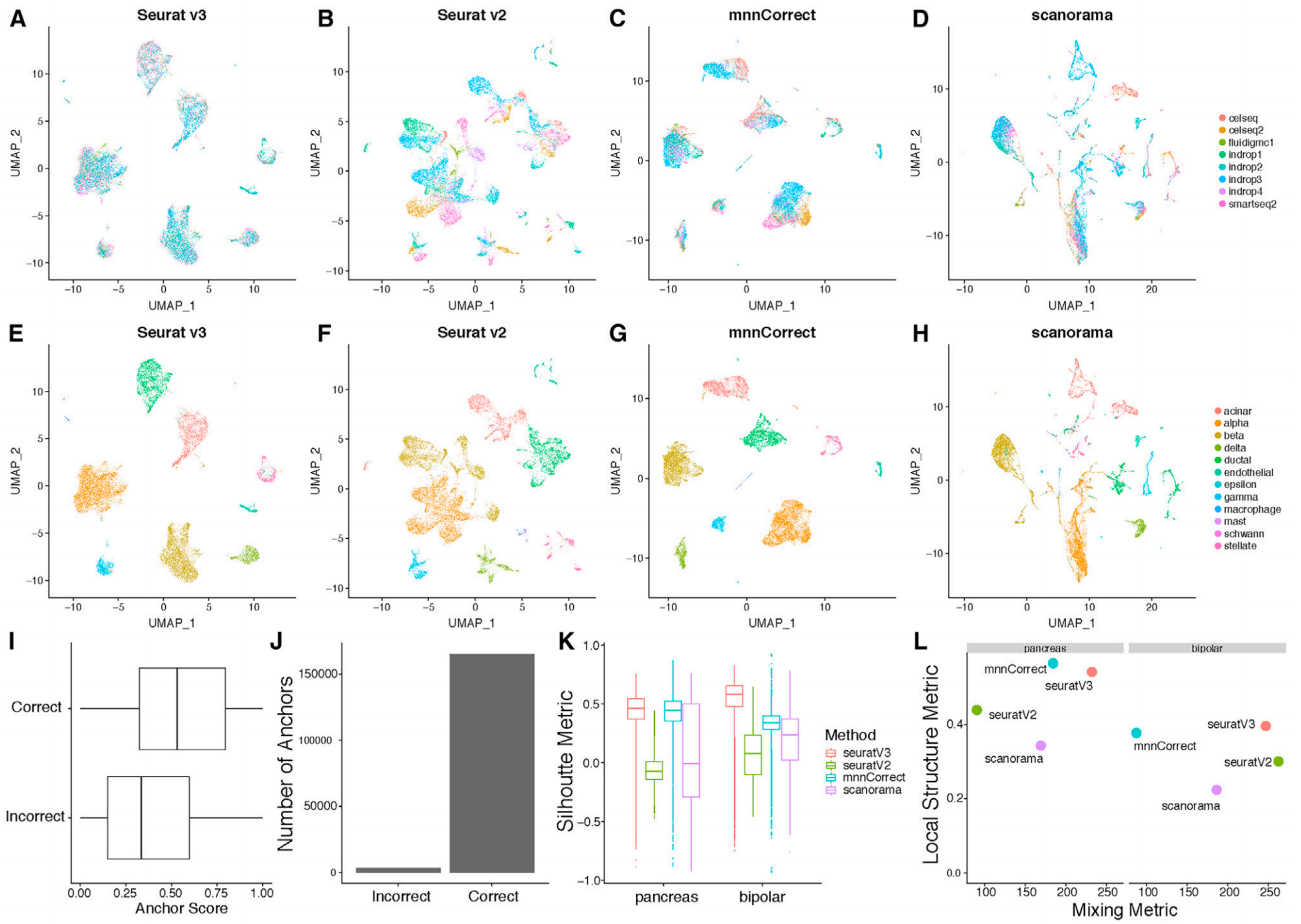
如图A-H展示了八个胰岛细胞数据集的 UMAP 图。[1]
参考文献
[1] Stuart T, Butler A, Hoffman P, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177(7):1888-1902.e21.