生信爱好者周刊(第 29 期):Hiplot开发库开源¶
这里记录每周值得分享的生信相关内容,周日发布。
本杂志开源(GitHub: openbiox/weekly),欢迎提交 issue,投稿或推荐生信相关内容。
封面图¶

Hiplot Gallery。via
本周话题:Hiplot开发库开源¶
Hiplot 项目发起于 2019 年,是由国内生物信息学开源社区 Openbiox 和多家单位和机构共同建设的一个免费、易用、部分开源的综合在线绘图系统(生物医学为主)。截至目前,该网站已提供超过 230+余个在线可视化分析功能,涵盖了基础科研绘图、组学可视化和部分临床模型可视化功能。总的注册用户已超过 2 万 5 千人,总访问量超过 300 万次,每日任务数已超 4000 余次。
近日,为了让更多生物信息学领域内的同道可以方便地在本地运行我们已开源的可视化工具(https://github.com/hiplot/plugins-open)以及为 Hiplot 网站贡献插件,我们开源了 Hiplot 网站应用的本地运行和开发库:https://github.com/hiplot/hiplotlib。如果你想为 Hiplot 网站添加颜色、主题或者部分新基础特性,都可以向这个仓库发起 Pull request 请求。
生信科技动态¶
1、Nat Biotechnol|基于多中心、多平台、高深度WGS的参考调用集,为癌症突变检测提供更高参照标准
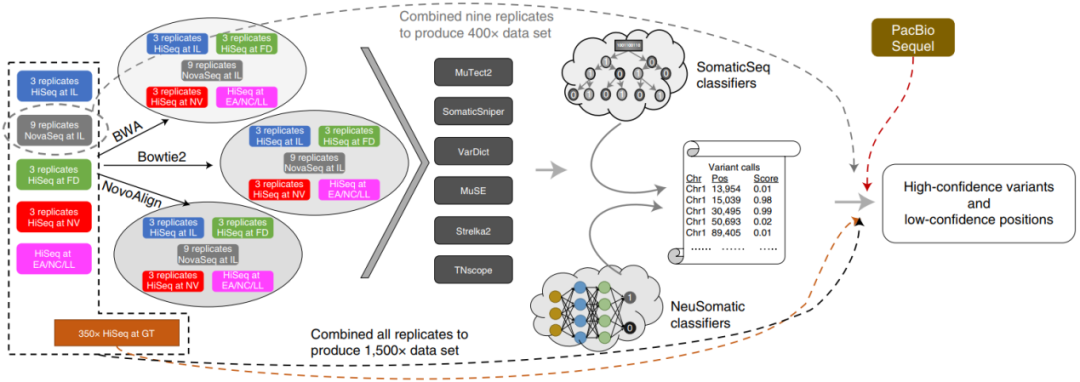
研究团队选取三阴性乳腺癌(TNBC)细胞系 (HCC1395)和B淋巴细胞衍生的正常细胞系(HCC1395BL),利用WGS跨越7个测序中心对配对肿瘤-正常细胞系的全基因组进行了深度测序(1500X),最大限度地减少了特定测序平台、检测中心或生物信息学算法的偏差,最终在HCC1395细胞系的整个基因组中创建了高可信度的突变调用数据,即“参考调用集”。此外,研究团队还通过靶向测序、单细胞DNA测序分析等方法对突变调用集进行了验证,提供了具有临床相关注释的高可信度种系和体细胞调用集。
2、Signal Transduction and Targeted Therapy | 癌症免疫研究的技术进步:从免疫基因组学到单细胞分析和人工智能

【综述】肿瘤细胞与附近的细胞一起存在于复杂的细胞群落中,这强烈影响肿瘤细胞的生长、行为和与其他细胞的交流。在这些细胞中,免疫细胞是关键的参与者,许多研究证明肿瘤细胞和免疫细胞之间的交流是双向的。事实上,免疫细胞既能促进也能抑制癌变、肿瘤进展、转移和复发。因此,文章主要关注肿瘤免疫微环境(TIME)。
3、Nature Communication | 北京大学席瑞斌组开发单细胞基因融合探测算法scFusion
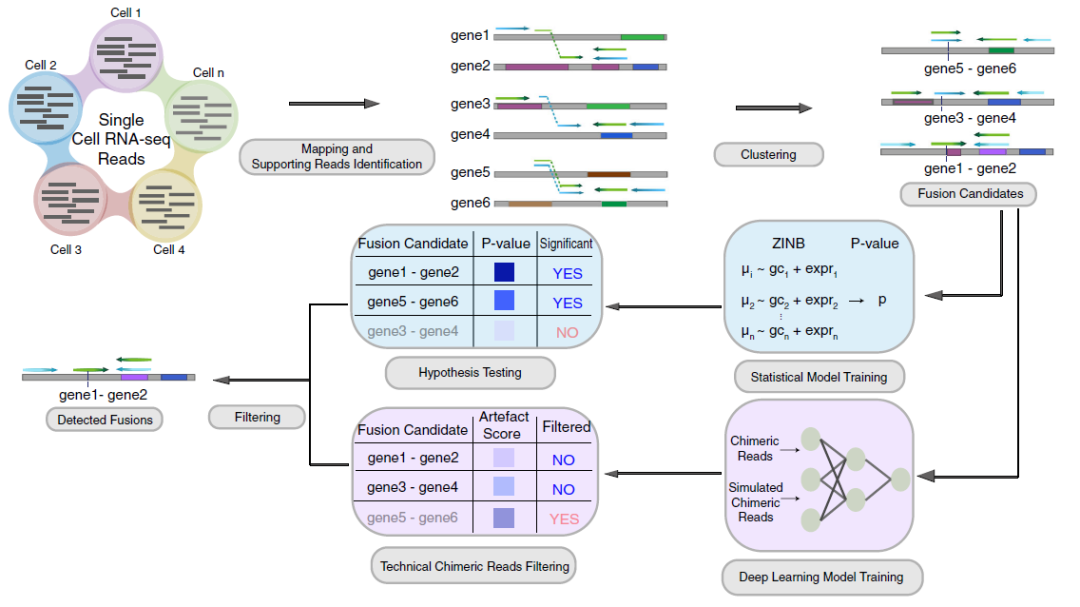
scFusion整合来自每个细胞的嵌合序列得到候选基因融合,再利用统计模型和深度学习模型从中筛选出较为可靠的结果。scFusion首先利用STAR进行序列映射,提取出嵌合序列并得到初始候选基因融合(几万个到几十万个)。由于真实的基因融合事件并不会太多,绝大部分候选基因融合都是技术原因造成的假阳性。席瑞斌团队基于广义可加模型(generalized additive model)对融合的支持序列数进行建模,从中估计出背景噪声的分布;再用统计假设检验方法,从中挑选出嵌合序列支持数显著高于背景噪声的基因融合。另一方面,部分技术原因造成的假阳性也会反复出现,且有非常高的嵌合序列支持数,统计模型中很难去除这些假阳性。为了进一步去除这些假阳性,scFusion通过构建bi-LSTM神经网络,学习由技术原因造成的嵌合序列的特征,从而去除相应的假阳性。经过两个模型的过滤,scFusion探测基因融合具有很高的灵敏度和特异度。
4、Genome Biology | 基于长读长转录组测序检测融合基因的新方法——JAFFAL
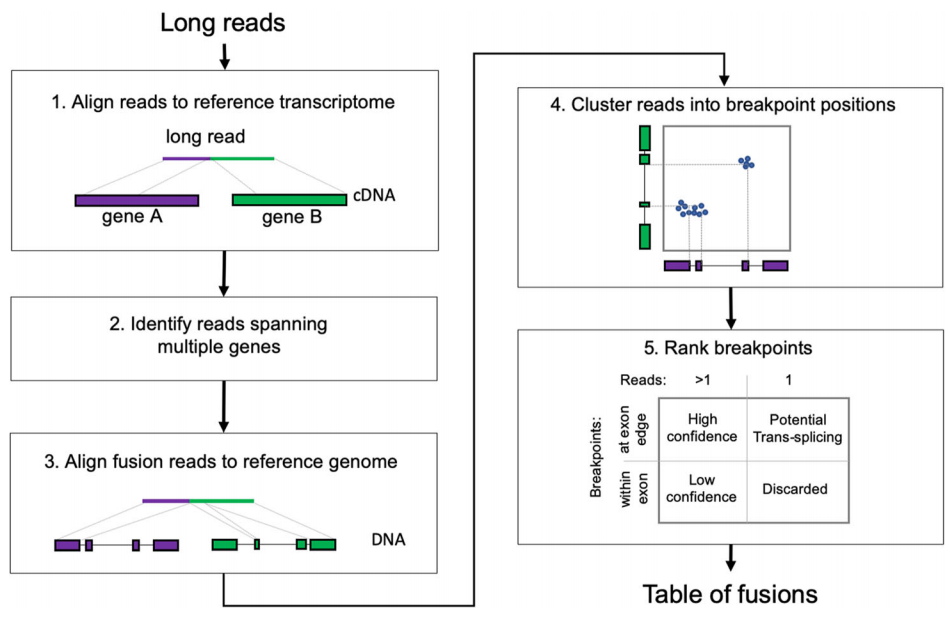
目前,只有三种融合基因检测方法可用于长读长转录组测序数据:JAFFA、Aeron、LongGF。JAFFA虽然可以处理任何长度的转录组测序数据,但测序错误率较高时,其灵敏度很低;Aeron是基于参考转录组将长序列片段比对图形上来检测融合基因。LongGF可分析基因组长读长测序数据,并通过识别与多个基因比对的reads来检测融合。
为了利用新的长读长测序技术进行融合基因发现和表征,研究团队基于此前开发的JAFFA研发了新的融合基因检测工具JAFFAL。JAFFAL是一种用 bpipe 编写的新多级计算框架。研究团队使用模拟数据、细胞系和来自ONT和PacBio的患者数据对JAFFAL进行了验证,并将JAFFAL应用到单细胞数据中,发现了跨越三个基因的融合,展示了从复杂重排中检测到的转录本。
文章¶
工具¶
1、toml-bench: 在python中该使用哪个toml包呢?
这个项目对python中不同toml包进行了测试,让人更全面的了解不同的toml包的优缺点。让人很容易选择适当自己项目的toml包。也让toml包开发者有了一套基准测试,让他们对自己的包的优缺点了如指掌并在后续进行针对性改进。
本文介绍一款完全免费的无需登录的pubmed插件,他可以解决pubmed本身不显示杂志影响因子的问题,重要的是他还能显示即时影响因子,中科院分区和杂志预警信息。
3、ask - Friendly CLI interaction in R

4、cmd - Code generator to produce CLI from R packages
5、Flux.jl - ML library that doesn’t make you tensor

Flux是一种优雅的机器学习方法。它是一个100%纯Julia的栈,在Julia本地GPU和AD支持的基础上提供了轻量级的抽象。Flux让简单的事情变得简单,同时保持完全可被黑客攻击。
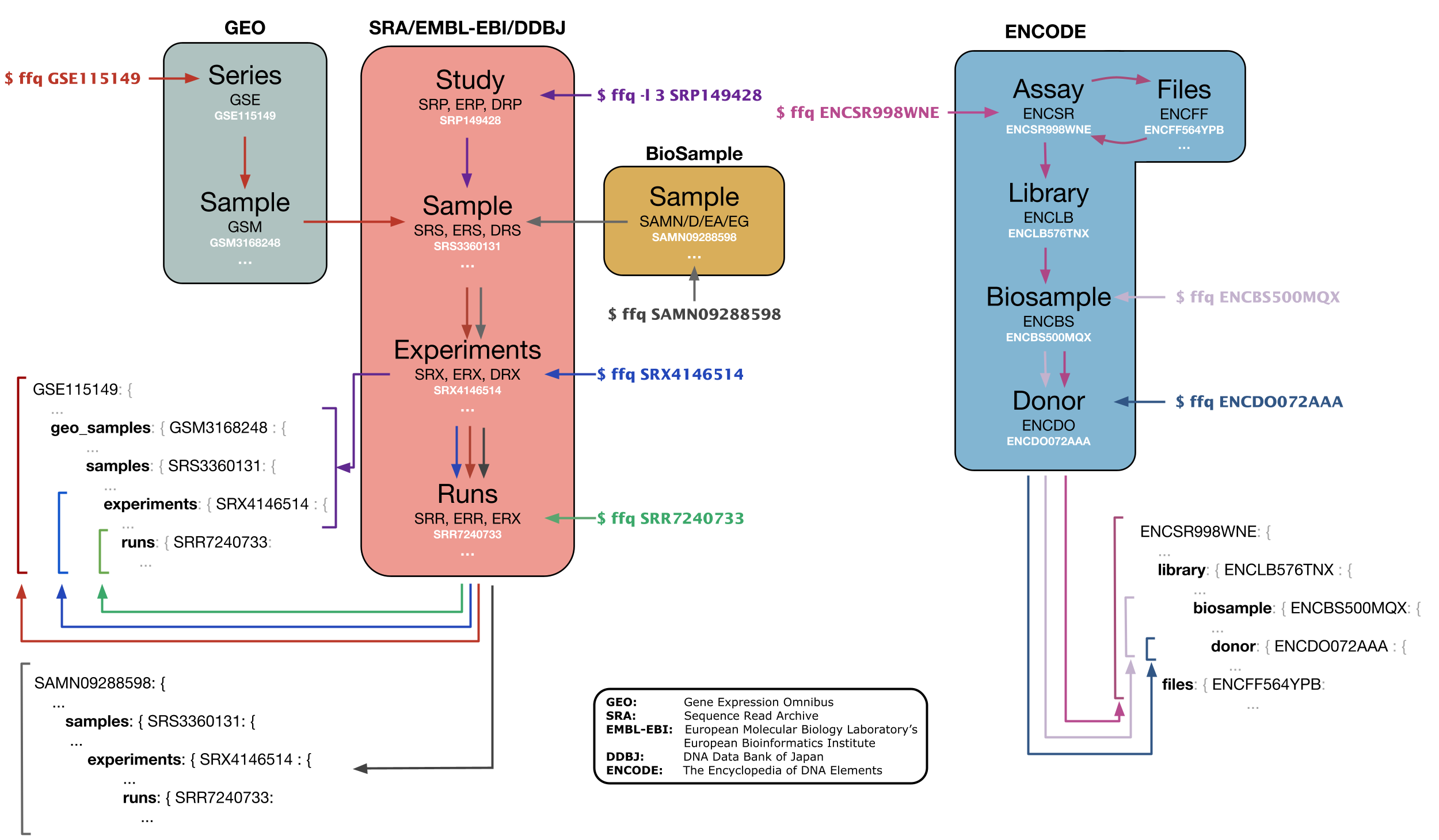
6、ffq - A tool to find sequencing data and metadata from public databases
(可以用于获取数据链接然后实现可编程下载)
从以下数据库中获取元数据信息:
- GEO: Gene Expression Omnibus,
- SRA: Sequence Read Archive,
- EMBL-EBI: European Molecular BIology Laboratory’s European BIoinformatics Institute,
- DDBJ: DNA Data Bank of Japan,
- NIH Biosample: Biological source materials used in experimental assays,
- ENCODE: The Encyclopedia of DNA Elements.
资源¶
DeeCamp2022 公益训练营面向所有计算机、生物、数学、物理、化学及其他生命科学相关交叉科学领域的本、硕、博在校生开放报名,全程免费。

DeeCamp2022 将通过线上方式进行,分为大师课和创新挑战赛两部分。成功加入 DeeCamp 的学员,将获得多位 AI 及生命科学领域的科研及产业顶级大师在线授课。
贡献者¶
赞赏¶
如果你想要支持本周刊,可以对推文进行赞赏或者提供的支付宝/微信二维码打赏。
感谢以下读者往期的赞赏:
- 成铮
订阅¶
这个周刊每周日发布,同步更新在微信公众号「生信协作组」(elegant-r)上。
微信搜索“生信协作组”或者扫描二维码,即可订阅。

(完)