生信爱好者周刊(第 110 期):至少20年不过时的研究方向在哪里?¶
这里记录每周值得分享的生信相关内容,周日发布。
本杂志开源(GitHub: openbiox/weekly),欢迎提交 issue,投稿或推荐生信相关内容。
封面图¶

本周话题:至少20年不过时的研究方向在哪里?¶
前不久,一位国内一流大学的青年博导作为委员,参加老文(笔者)学院一个博士生的学位论文答辩会。答辩会结束后,几位导师喝茶闲聊,青年博导谈到自己的一个研究生给他提出选题的要求:希望研究方向至少20年不过时。青年博导说他不知道这样的方向在哪里。
老文第一时间对青年博导表达由衷羡慕之情,羡慕他遇到了一个千载难逢的好学生。
为什么说这个学生千载难逢?至少20年不过时的研究方向又在哪里?
生信研究¶
ecDNA是近年来引起广泛关注的一种肿瘤基因组结构异常,超过14 %的原发性肿瘤和40 %的转移性肿瘤存在ecDNA扩增。ecDNA游离于染色体外,呈环状,且具有自我复制功能。其含量与耐药性呈正相关,是肿瘤基因组学研究的新方向,也是极具潜力的诊断标志物。目前除了将全基因组测序(WGS)、高通量染色质空间构象捕获测序(Hi-C)、三代测序(Third-generation sequencing)三种方法联合解读的策略,没有很好的办法对ecDNA的序列信息以及三维空间构象信息进行解读。
研究团队开发了一种新型多重基因组结构变异检测方法 - MGA-Seq(Multiple genetic abnormality sequencing)。该方法充分利用ecDNA在细胞核中游离的特性,首先将ecDNA与邻近的染色质进行交联,再利用限制性内切酶和连接酶对肿瘤染色质进行原位片段化、邻近酶连,将ecDNA片段与相邻的基因组DNA片段连接在一起。最后,将连接后的片段进行超声片段化,构建成测序文库。通过对反应体系的优化,研究人员将MGA-Seq所有的反应步骤合并到一个离心管中,只需9小时、花费56美金即可完成文库构建步骤,并实现对复杂基因组结构异常的精准检测。
-
论文链接:https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03081-x
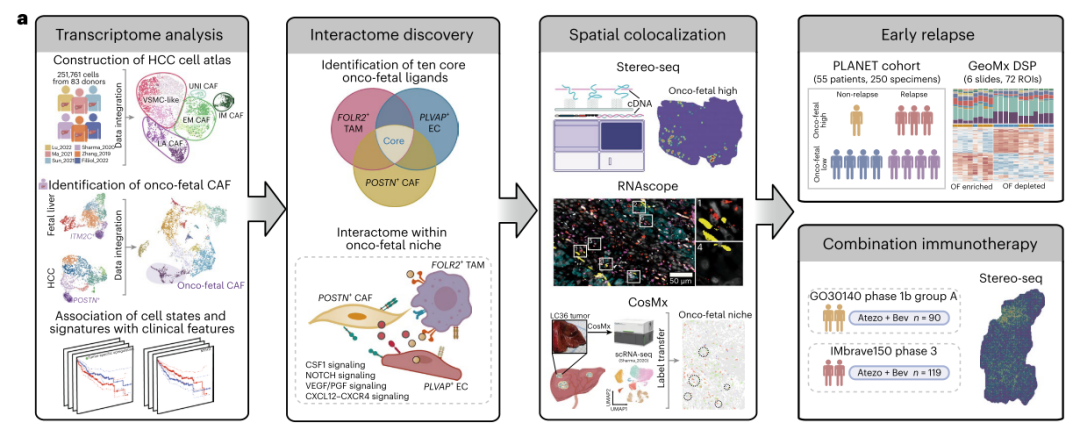
近日,上海市免疫学研究所Florent Ginhoux研究团队、澳大利亚科廷大学Ankur Sharma团队及新加坡国家癌症中心Pierce Kah-Hoe Chow团队,使用华大自主研发的时空组学技术Stereo-seq,并结合scRNA-seq和bulk RNA-seq,系统地描绘了肝细胞癌(hepatocellular carcinoma,HCC)肿瘤相关成纤维细胞(cancer-associated fibroblasts,CAF)图谱及胎肝成纤维细胞图谱,首次鉴定出了一群参与HCC“癌-胚”重编程(onco-fetal reprogramming)过程的重要细胞类群POSTN+ CAF,并证明了POSTN+ CAF、FOLR2+ TAM和PLVAP+ EC这三群“癌-胚”细胞的空间共定位关系,揭示了“癌-胚”重编程过程对HCC患者术后复发及治疗耐药的影响,为HCC的分层诊疗及开发新的免疫治疗策略提供了重要的分子靶点。
-
论文链接:https://www.nature.com/articles/s43018-023-00672-2

小RNA广泛存在于植物中,对植物的生长发育和营养繁殖起至关重要的作用。目前已有多款植物小RNA分析工具,但大多数依赖于命令行环境,极大阻碍了湿实验室生物研究人员在植物小RNA生物功能挖掘上的深度探索。此外,当前的小RNA分析工具多集中在单一类型的小RNA分析上,且计算资源消耗较大,通常需要大量时间和精力来学习众多工具或脚本的使用并组成可用的流程以获得最终分析结果。
2023年12月30日,华南农业大学夏瑞团队在Science Bulletin(IF:18.9)上在线发表了题为“sRNAminer: a Multifunctional Toolkit for Next-Generation Sequencing Small RNA Data Mining in Plants”的研究论文,该研究开发了一款多功能植物小RNA分析工具——sRNAminer。利用该工具,研究人员可进行一站式小RNA分析及可视化。
-
论文链接:https://www.sciencedirect.com/science/article/abs/pii/S2095927323009350
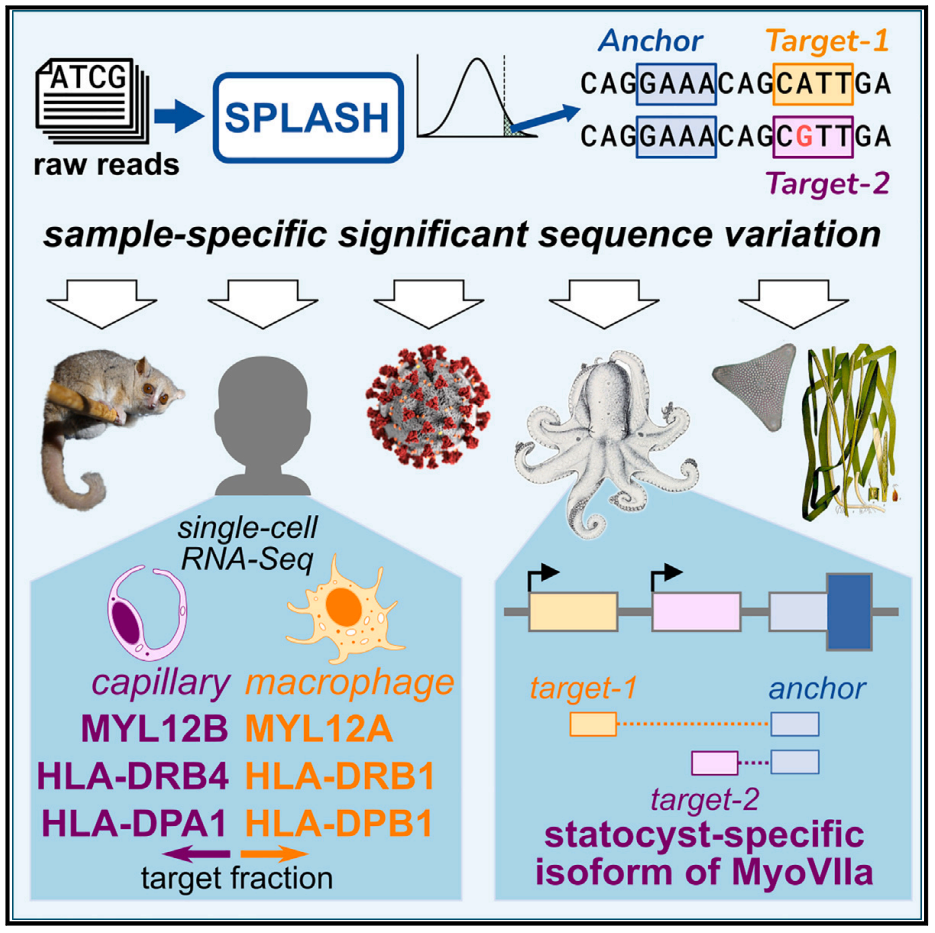
基因组学与生物学、生态学和医学有着密不可分的关联。近年来,随着测序数据的海量增长,如何才能更好地分析这些数据,揭示其规律和功能成为了迫切需解决的问题。基因组学的工作流程通常先将测序数据读取映射到参考基因组上,从而进一步展开下游分析。参考基因组是生信分析的基础,影响着下游分析的质量。参考基因组的缺失或不完整将极大的限制生物学发现。与参考基因组对齐这一步骤大大影响了测序数据分析的准确度与计算性能的敏捷性。同时,生物的多样性对对齐参考基因组以及下游生物学发现有着许多直接或间接的影响。
近日,来自美国斯坦福大学的Julia Salzman团队Cell 期刊上发表题为SPLASH: A statistical, reference-free genomic algorithm unifies biological discovery的文章。研究人员构建了一个新的、普适性强的高效统计算法——SPLASH。SPLASH 可直接从原始测序数据进行序列变化的统计驱动分析,克服了以往基因组学数据分析中对参考基因组的依赖,计算效率高,可大规模运行以检测各种形式的样本特异性序列差异。
- 论文链接:https://www.cell.com/cell/fulltext/S0092-8674(23)01179-0?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS0092867423011790%3Fshowall%3Dtrue
博文资讯¶
https://mmbiz.qpic.cn/mmbiz_gif/qEtdU1oebKM6Wzj7Yr7uXFuKREs02LmUdIxMwiax7AiajqbYsVMCibAPIconqBFkJibkW3HkhVBPibdicqK7dqGDUibkw/640?wx_fmt=gif&from=appmsg&wxfrom=5&wx_lazy=1
Linux 是怎么启动的?
几乎每个软件工程师都用过 Linux,但并不是每个人都知道它的启动过程。让我们深入了解一下。
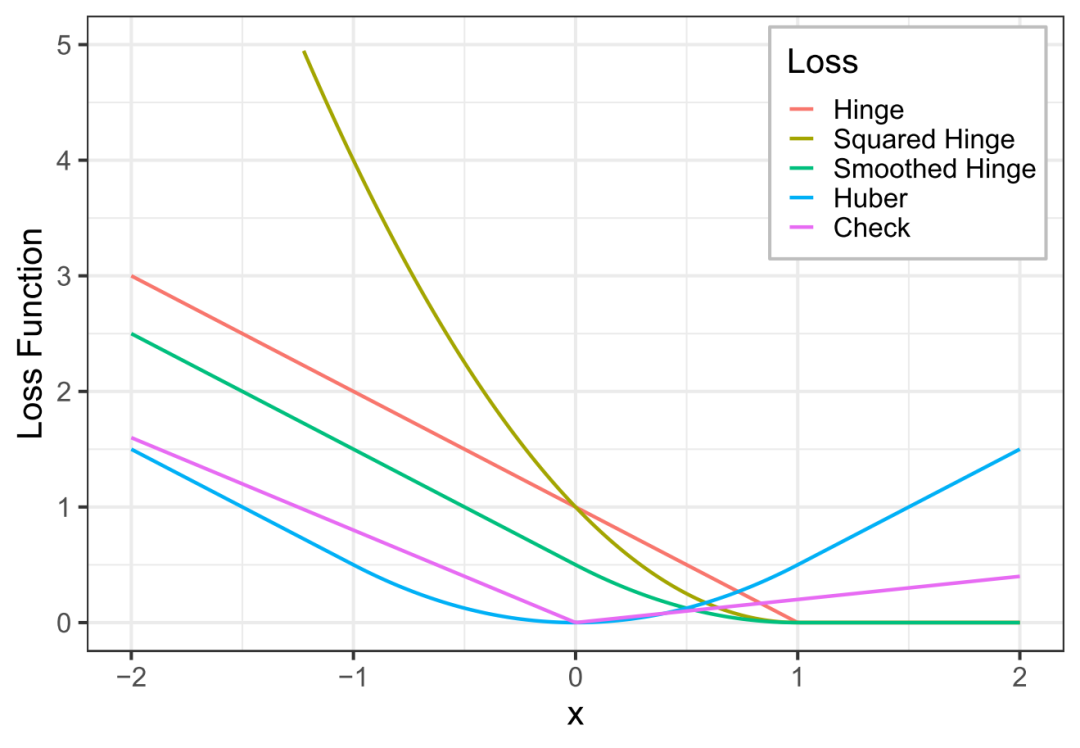
长久以来,机器学习江湖中一直流传着两件神器——LibSVM 和 Liblinear,它们都是用来求解支持向量机(SVM)问题的,且都是由台湾大学的林智仁教授及其团队开发和维护。两者的不同在于,LibSVM 支持各类基于核函数的 SVM,而 Liblinear 只能计算线性 SVM。但从计算效率上来说,LibSVM 随样本量 n 增加的复杂度大约在 $\ce{O(n^2)}$ 到 $\ce{O(n^3)}$ 之间,而 Liblinear 则几乎是线性的复杂度 $\ce{O(n)}$ 。因此,如果考虑使用线性模型,那么 Liblinear 基本可以说是求解 SVM 的性能天花板。
那么 Liblinear 这柄屠龙宝刀是否就从无敌手,一直号令天下呢?属于 SVM 江湖的倚天剑又流落何方?我们的故事,便由此说起。
本文指导初学者开始深度学习,对诸多基础概念和框架进行了介绍。
本文介绍了如何从最新的Seurat v5版本降级到v4版本的方法。
工具¶
CZ CELLxGENE Annotate是一个用于单细胞数据集的交互式数据浏览工具,可以对来自人类细胞图谱的数据集实现至少100万细胞的快速可视化。
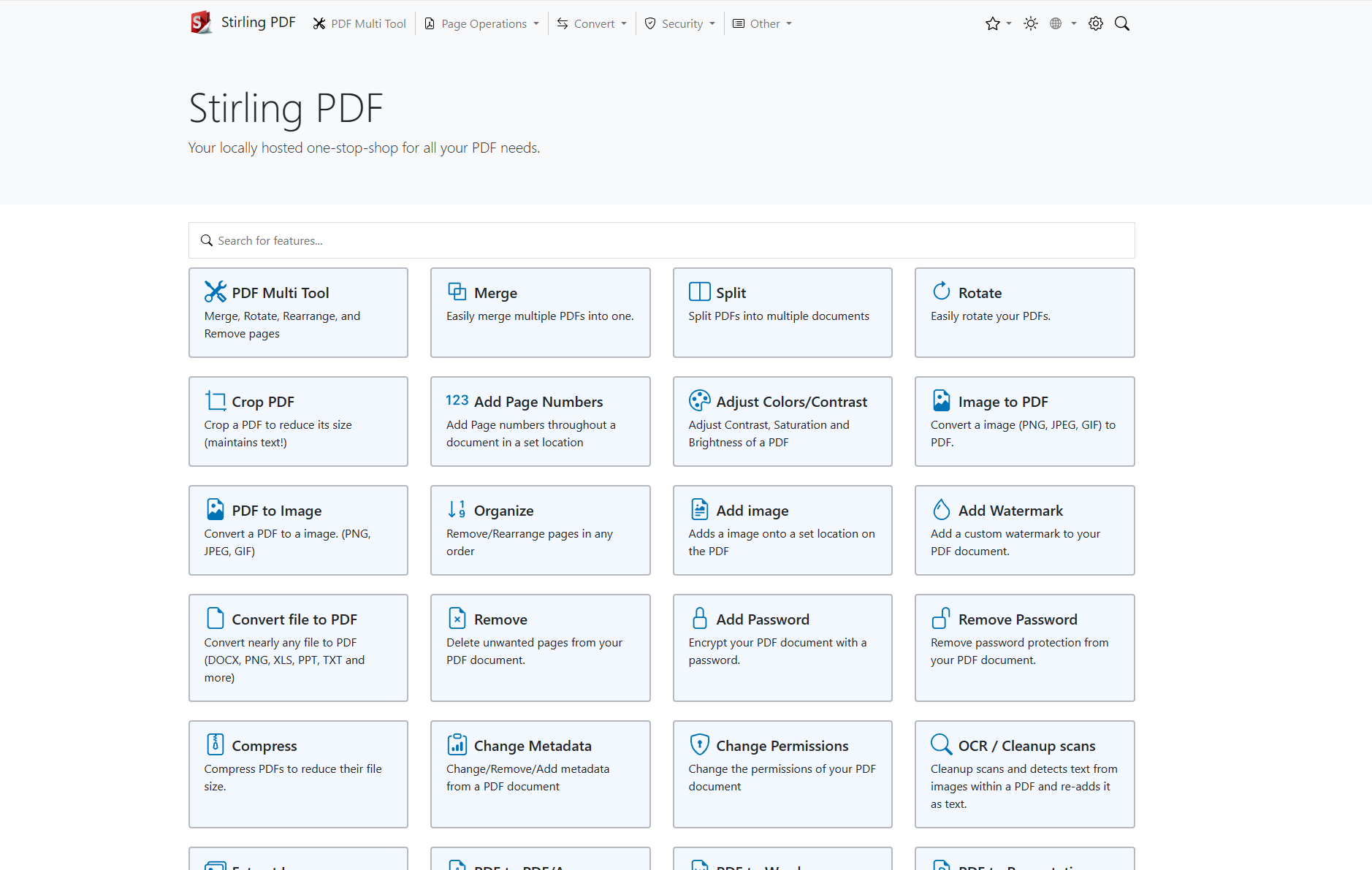
Stirling-PDF是一款强大的本地托管的基于Web的PDF操作工具,借由docker允许用户对PDF文件执行各种操作,例如拆分合并、转换、重组、添加图像、旋转、压缩等。最初,这个本地托管的Web应用程序是一个100%由的ChatGPT制造的应用程序,现在发展出更广泛的功能,以满足用户对PDF的各种需求。
资源¶
本文介绍了肿瘤免疫机制、CAFs、CD4+T、泛素化、T细胞应激反应状态、癌症的起源、中性粒细胞、PD-1和T细胞、肿瘤微环境的动态变化、组织驻留免疫细胞这十个方向的高分综述。
历史上的本周¶
贡献者(GitHub ID)¶
「Openbiox 生信周刊」运维小队:
@ShixiangWang(王诗翔)@kkjtmac(阚科佳)@NiEntropy(赵启祥)@He-Kai-fly(何凯)@JnanZhang(张佳楠)@Tomcxf(陈啸枫)@wangdepin(王德品)@kongjianyang(空间阳)
订阅¶
这个周刊每周日发布,同步更新在微信公众号「生信协作组」(elegant-r)上。
微信搜索“生信协作组”或者扫描二维码,即可订阅。

(完)