生信爱好者周刊(第 121 期):不唯论文,在中国可行吗?¶
这里记录每周值得分享的生信相关内容,周日发布。
本杂志开源(GitHub: openbiox/weekly),欢迎提交 issue,投稿或推荐生信相关内容。
封面图¶

- 图示:幻想未来的生物技术。(由AI生成)
本周话题:不唯论文,在中国可行吗?¶
饶毅:科学研究当然有论文,“不唯论文”的意思是不仅仅看论文数量、论文在哪里发表,而看论文研究的问题是否重要、研究内容是否有科学意义或应用价值。
-
科学杂志主编霍尔顿·索普:我认为这个问题的根源与我之前所说高等教育竞争程度以及机构之间的相互竞争有关。竞争在某些方面是好的,对于大型研究机构来说,他们不断追逐各种排名,追逐从国家科学基金获得多少资金,并希望把所有这些放在他们的网站和宣传册上,犹如宣传自己的业绩一样。这种文化就是这样创造滋生而来,如明星教师被招聘的依据是他们所获的资金和他们在高知名度的出版物。在这整个过程中,教育被忽略了,它是整个事件中最不幸的牺牲品。科学和教育应该是有灵魂的东西,用量化非人性化的冷酷竞争模式,是现代科学出版的根源所在。
-
@ShixiangWang(王诗翔):个人看来,不唯论文和绩效的量化在资源竞争激烈的当下是不可调和的矛盾,短期内无法实现。
生信研究¶
1、Cell | 崔亚/李蔚团队利用33万全基因组测序数据揭示人类跨族群串联重复序列扩张的遗传奥秘
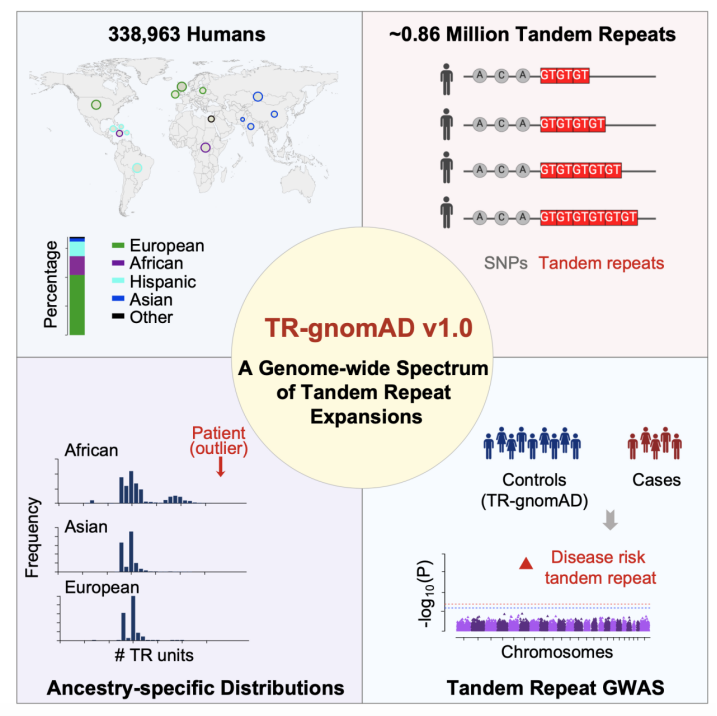
2024年4月5日,美国加州大学(尔湾)研究助理教授崔亚和讲席教授李蔚实验室在Cell杂志上发表文章A genome-wide spectrum of tandem repeat expansions in 338,963 humans,该研究全面分析了33.9万人类全基因组测序数据,建立了第一个Biobank级别的人类多种族串联重复序列扩张遗传变异谱TR-gnomAD,促进TR expansions在人类遗传疾病中的研究。
- 论文链接:https://doi.org/10.1016/j.cell.2024.03.004
2、Advanced Science | 高远/赵方庆合作研发单细胞和空间转录组中环形RNA深度学习算法

本研究研发了深度学习模型CIRI-deep,以准确预测不同样本间的差异剪接环形RNA。该模型从环形RNA调控机制角度出发,整合了3527个环形RNA特异的顺式元件以及1499个样本特异的反式因子作为输入特征,且不依赖于传统的反向剪接信号识别,可以在任意转录组样本间预测差异剪接的环形RNA。评估结果表明,CIRI-deep可以实现多种转录组测序数据中差异剪接环形RNA的可靠预测,并在单细胞及空间水平实现细胞类型特异环形RNA的准确解析,具有广泛的应用场景。
- 论文链接:https://doi.org/10.1002/advs.202308115
3、Nature丨王红阳/白凡/陈磊/Steven Rozen团队联合揭示中国人群肝癌全基因组变异及演化特征


2024年2月14日,海军军医大学王红阳院士/陈磊研究员课题组联合北京大学生物医学前沿创新中心/北京未来基因诊断高精尖创新中心白凡教授课题组、杜克-新加坡国立大学医学院Steven G. Rozen教授课题组以及福建和瑞基因吴琳博士团队合作在Nature在线发表了题为 Deep whole-genome analysis of 494 hepatocellular carcinomas 的最新研究成果,完成了中国人群肝细胞癌全基因组深度特征分析。本研究对494例来自中国不同地区的肝细胞癌患者肿瘤组织进行了高深度的全基因组测序(平均120x),深入分析了编码区和非编码区的驱动基因、突变印记、拷贝数变异、聚集式变异事件、染色体外环状DNA以及突变演进规律等特征,揭示了以HBV相关肝细胞癌为主的全基因组变异景观,为深入理解中国人群肝细胞癌的演进机理提供了重要的线索。
- 论文链接:https://www.nature.com/articles/s41586-024-07054-3
4、Nature Biotechnology | 基于时空单细胞的生成模型,揭示细胞-药物干扰反应和感染的动态轨迹

Biolord模型,是Mor Nitzan团队开发的一个前沿的深度生成框架,旨在通过输入单细胞转录组数据,解构和解码细胞类型、组织来源和分化阶段等复杂属性。该研究发表于2024年1月的《Nature Biotechnology》期刊,标志着单细胞数据科学的重要进展。面对单细胞数据中多重属性解析的挑战,Biolord通过其创新的建模方法独立解码每个属性,并利用细胞的潜在表示进行测量预测,显著提高了对药物和遗传干扰反应的预测准确性。该工具不仅为生物医学研究提供了新的视角,也为个性化医疗和精准治疗的发展提供了强大的分析手段。
- 论文链接:https://www.nature.com/articles/s41587-023-02079-x
博文资讯¶
5、第一作者解读 | 我们这篇广州出生队列基因组学研究的 Nature 文章都讲了什么

2024 年 1 月 31 日,笔者作为第一作者的广州出生队列基因组学研究的文章——“The Born in Guangzhou Cohort Study enables generational genetic discoveries”——正式在 Nature 主刊上发表了,这是广州出生队列第一个里程碑意义的成果。在这篇推文中,笔者对这项工作做了更专业、深入和完整的解读,讲述了这项研究的具体发现,以及项目过程中的一些思考和体会。
- 原文链接:https://www.nature.com/articles/s41586-023-06988-4
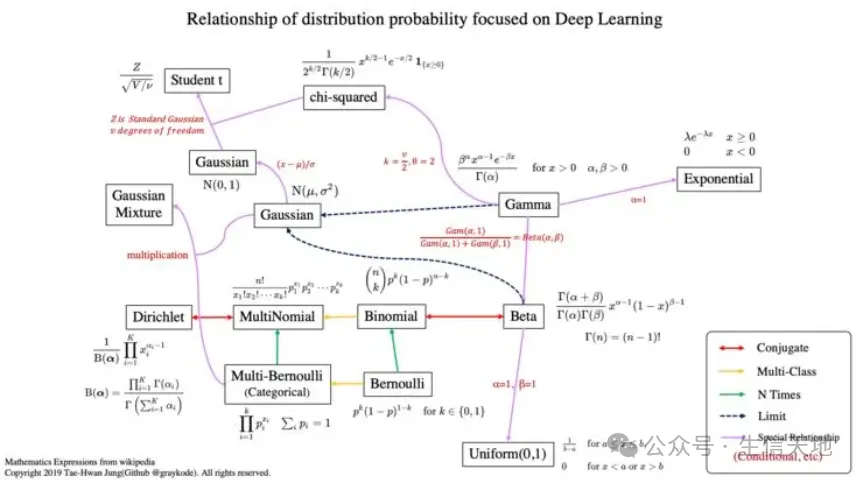
生物信息学是一门跨学科的科学,涉及生物学、计算机科学、信息工程、数学和统计学等领域的知识。在生物信息学中,概率分布是一种重要的工具,用于描述和分析生物数据。本文将对生物信息学中常见的概率分布进行综述,包括离散分布、连续分布、抽样分布以及其他分布,并讨论它们在生物信息学中的应用。

这篇文章介绍了中国近年来新兴的一批高等教育机构和科研机构,它们与国家高等教育革新和科技创新紧密相关,展现出活力和创新精神。文章特别提到了西湖大学、深圳医学科学研究院、首都医科大学创新研究中心、宁波东方理工、福耀科技大学、深圳理工大学、大湾区大学和上海科技大学等机构,并探讨了它们成为世界顶尖科研中心的潜力。
本文介绍一种为了T2T基因组整套组装流程而去建自动化流程工具:quarTeT( Telomere-to-telomere Toolkit)。quarTeT实际上共有四个模块:AssemblyMapper、GapFiller、TeloExplorer和CentroMiner,分别对应基因组的组装,gap填充,端粒查找和着丝粒查找。
工具¶

一系列R 函数,用于拆分串联数据、堆叠数据集的列以及将数据转换为不同的形状。
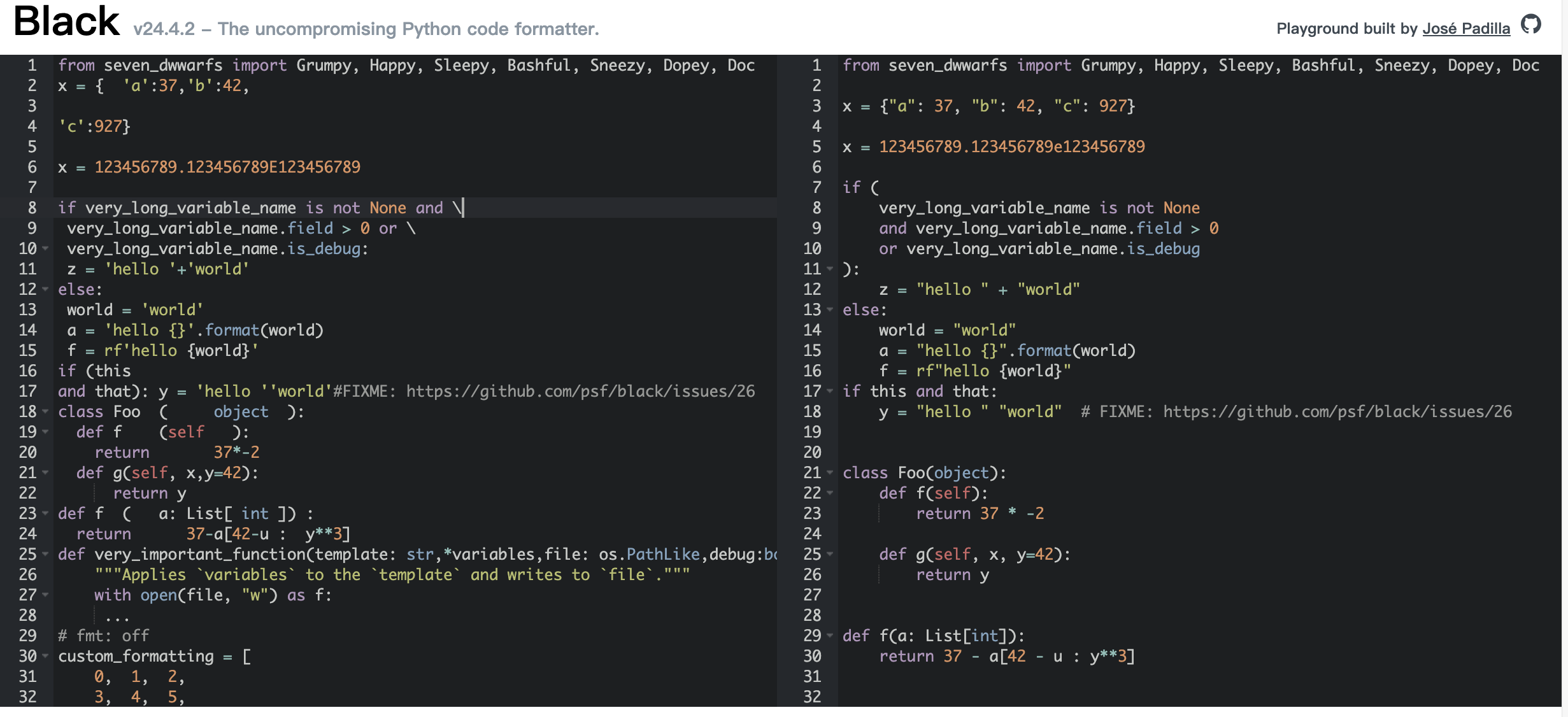
Black 是一个不妥协的 Python 代码格式化工具。使用它,你同意放弃对手工格式化细节的控制。作为回报,Black 提供了速度、确定性,并让你从 pycodestyle 对格式的唠叨中解放出来。你将节省时间和精神能量,专注于更重要的事情。无论阅读哪个项目,经过 Black 处理的代码看起来都是一样的。过一段时间,格式化变得透明,你可以专注于内容。Black 通过生成尽可能小的差异,使代码审查更快。
- Playground: https://black.vercel.app/
资源¶
清华大学生物信息学课件和实践教程。
12、Compare Seurat or Scanpy for single-cell RNA-seq analysis
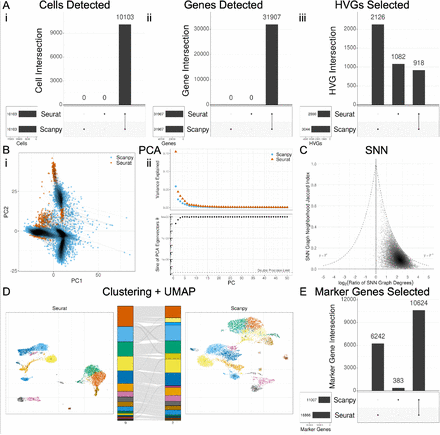
Seurat vs. Scanpy单细胞转录组分析比较的源代码。大家可以跑跑试试,同样的单细胞数据,可能得到的结果不一样。
13、Great Resources for Learning Generative AI and Large Language Models
收集多个学习资源来理解LLMs。想了解LLM的同学可以看看。
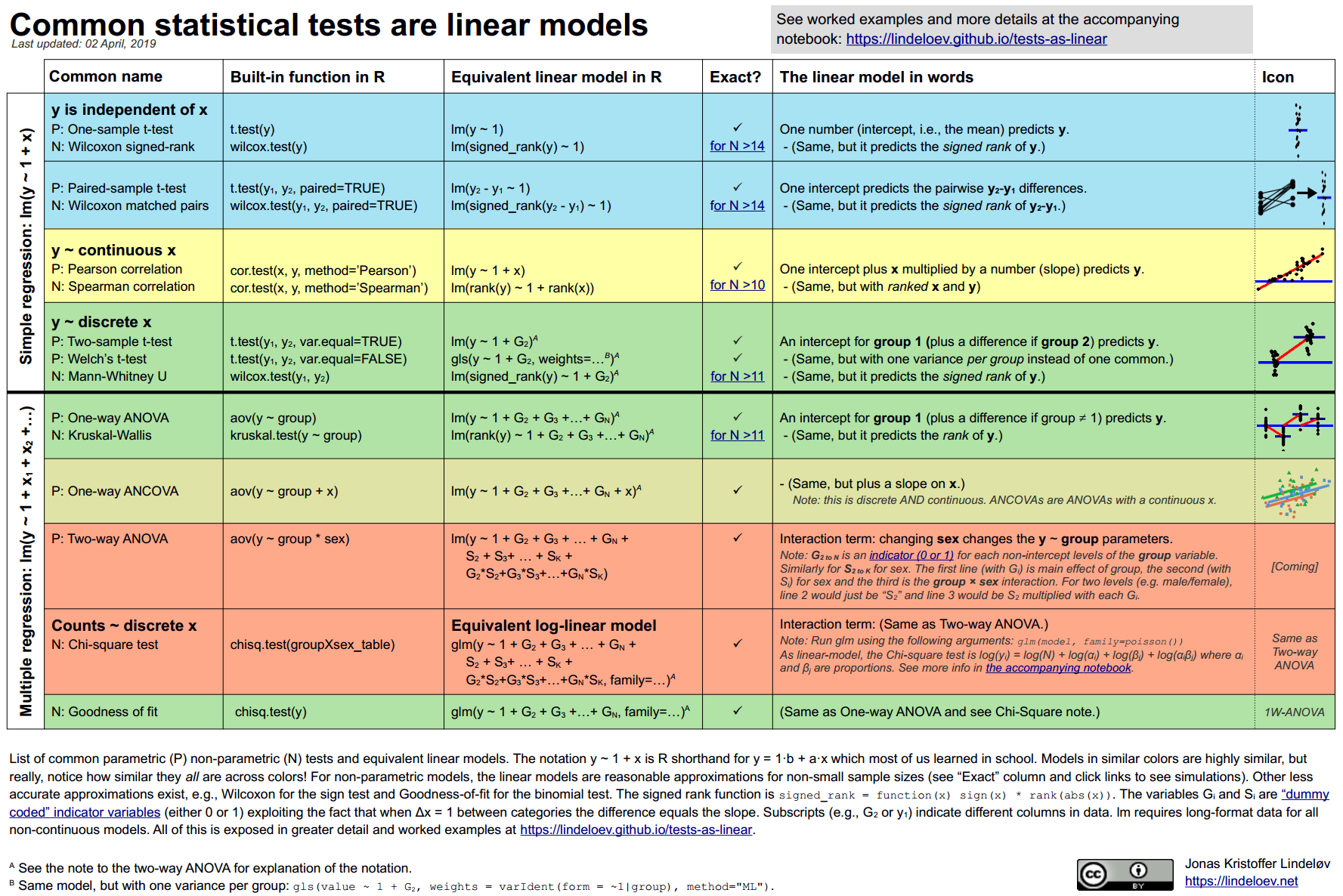
本文总结了常见参数和“非参数”测试的线性模型。
历史上的本周¶
贡献者(GitHub ID)¶
「Openbiox 生信周刊」运维小队:
@ShixiangWang(王诗翔)@kkjtmac(阚科佳)@NiEntropy(赵启祥)@He-Kai-fly(何凯)@JnanZhang(张佳楠)@Tomcxf(陈啸枫)@wangdepin(王德品)@kongjianyang(空间阳)@donghongyu2020- 董弘禹
订阅¶
这个周刊每周日发布,同步更新在微信公众号「生信协作组」(elegant-r)上。
微信搜索“生信协作组”或者扫描二维码,即可订阅。

(完)