生信爱好者周刊(第 79 期):四千周¶
这里记录每周值得分享的生信相关内容,周日发布。
本杂志开源(GitHub: openbiox/weekly),欢迎提交 issue,投稿或推荐生信相关内容。
封面图¶

本周话题:四千周¶
一篇名为《反生产力宣言》的文章讨论了生产力的概念及其潜在的陷阱。作者引用了奥利弗-伯克曼(Oliver Burkeman)的《四千周》一书,该书强调了我们生活中有限的时间,并批评了对效率的无情追求。作者认为,高效往往会导致更多工作而不是更多闲暇时间,因为其他人开始期望你做得更多。这是由于工作量本质上是无限的。作者使用一个商人和渔民的寓言来说明追求效率以享受生活时的讽刺之处,当时生活中令人愉快的部分通常不需要效率。文章还讨论了被落下或变得无关紧要的恐惧,这影响着每个人,无论他们成功与否或财富如何。这场持续不断地竞赛经常导致遗憾,正如垂死之人普遍后悔过度劳累一样。作者建议解决这种生产力跑步机问题的方法是积极选择对你重要并花时间去做那些事情,无论是友谊、爱好还是健康等方面。作者还警告不要陷入沉迷于工作行为而非工作本身的陷阱。文章最后强调了珍视生命有限和我们所做选择的重要性,以及需要欣赏当前时刻而不是不断追求更美好未来的必要性。
@wangdepin - 很有意思的一点:高效率的人往往会得到更多的工作,而不是更多的休闲时间,因为其他人开始期待你能做更多的事情。
生信研究¶
1、Cancer Cell | d-MMR/MSI-H 肠癌免疫微环境解析
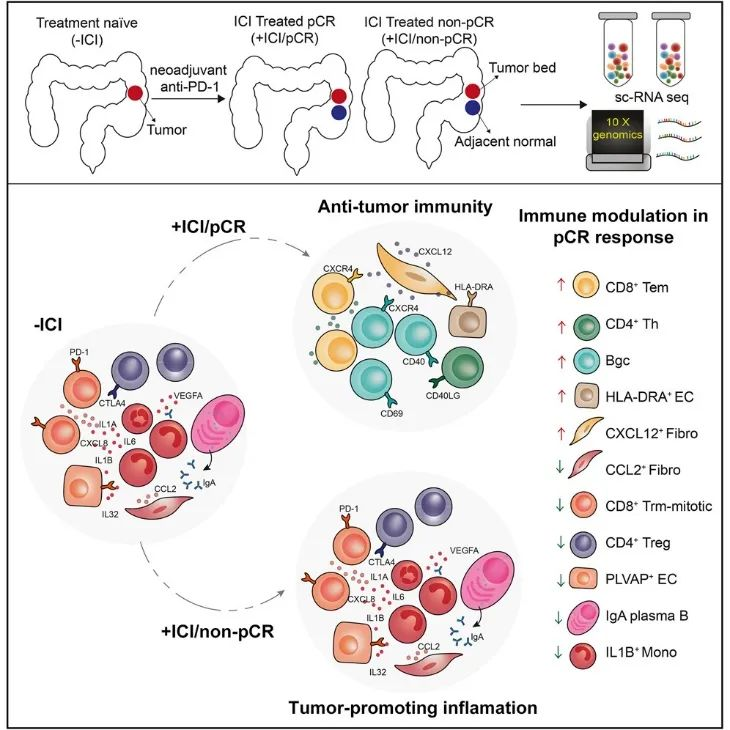
免疫检查点抑制剂(ICI)治疗可以诱导错配修复缺陷和微卫星不稳定性高(d-MMR/MSI-H)结直肠癌(CRC)的完全缓解。然而,免疫治疗实现病理完全反应(pCR)的潜在机制尚未完全了解。该研究首次对PD-1阻断剂(托利帕单抗)或托利帕单抗联合COX-2抑制剂(塞来昔布)治疗的d-MMR/MSI-H CRC患者的肿瘤细胞群进行了深入的细胞和分子分析。该研究旨在揭示肿瘤残余患者对ICI治疗的耐药性和敏感性的基础,并提示ICI治疗的潜在治疗靶点。该研究比较了实现病理完全缓解(pCR)的患者暴露于ICI前后的细胞类型分布和功能变化,以阐明与ICI治疗成功相关的机制(−ICI vs +ICI/pCR)。该研究还分析了pCR和非pCR反应在治疗动力学上的差异,以探讨免疫治疗耐药的机制。
- 文章链接:https://www.cell.com/cancer-cell/fulltext/S1535-6108(23)00137-X#%20
2、NEJM | 英国“破译发育障碍”项目揭示五千余例罕见病的遗传原因
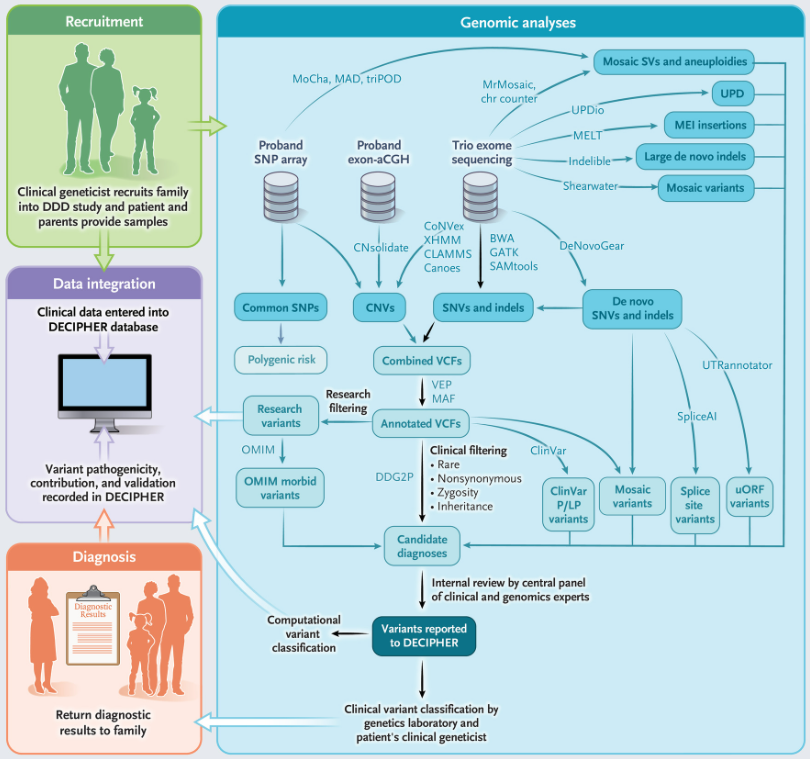
近年来,基因组测序在识别罕见单基因疾病的分子机制方面取得了非凡的进展,并越来越多地应用于多种疾病的诊断。最早将大规模基因组研究与个体患者反馈相结合的研究之一是英国破译发育障碍(Deciphering Developmental Disorders, DDD)研究项目。该项目招募了13500多个家庭,获得了外显子组测序和微阵列数据,并与200多名临床医生记录的丰富临床表型相补充。
近日,该项目团队在国际顶级期刊The New England Journal of Medicine发表了题为“Genomic Diagnosis of Rare Pediatric Disease in the United Kingdom and Ireland”的文章。在该文章中,研究团队描述了DDD研究多年来开发的分析策略。通过该项目,大约5500例严重发育障碍患者了解了其疾病相关遗传因素。平均而言,每个父母-后代三人组中有1.0个候选变异,每个单例先证者有2.5个变异。通过使用临床和计算方法进行变异分类,约41%的先证者(13,449人中有5502人)得到诊断,其中76%的个体携带致病性新生突变。另外22%的先证者(13449人中的2997人)在与单基因发育障碍密切相关的基因中存在意义不明变异。
- 论文链接:https://www.nejm.org/doi/full/10.1056/NEJMoa2209046

肺癌是最常见的恶性肿瘤之一。近年来,肺癌的全球发病率及死亡率均以惊人的速度上升。非小细胞肺癌(NSCLC)是最常见的肺癌类型,约占所有病例的85%。了解肺癌的特征变化可以揭示肿瘤如何演变,进而帮助开发新的更有效的治疗方法。
近日,发表在《自然》与《自然医学》上的TRACERx研究系列文章中,TRACERx联盟团队描述了癌细胞DNA的变化如何帮助研究人员预测细胞未来的行为,包括癌症将在何时何地转移扩散到身体的其他部位。研究团队从基因组、转录组、细胞、组织以及临床表现等不同层面对肺癌的进化和转移、异质性等关键癌症特征进行了表征,揭示了基因组变异在不同的肿瘤克隆演化模式和转移中的作用。
- 论文链接:https://www.nature.com/articles/s41586-023-05783-5
4、Nat Mach Intell | 彭绍亮教授课题组在基于多模态网络表征学习的药物发现方法研究中取得重要进展
近日,湖南大学信息科学与工程学院彭绍亮教授课题组在国际顶级期刊Nature Machine Intelligence发表了题为“Multi-task Joint Strategies of Self-supervised Representation Learning on Biomedical Networks for Drug Discovery”的研究论文。该研究提出了面向药物发现的生物网络自监督表征学习的多任务联合框架MSSL2drug,系统地研究了如何有效地组合多个自监督模型这一人工智能领域的挑战性问题,探索发现了基于生物网络的多模态网络表征学习技术,为缺乏生物或临床标注数据的药物发现提供了新的研究思路。

- 论文链接:https://www.nature.com/articles/s42256-023-00640-6
博文资讯¶
5、多分享

华南农业大学夏瑞老师写给学生的建议,公众号还有其他的文章。能感受到夏瑞老师是一个特别爱分享的人,给我们这些初入科研的学生很多诚恳的建议,很希望这样的老师多一点。
6、白话孟德尔随机化
本文用通俗易懂的语言解释了什么是孟德尔随机化及其研究相关要素,在文末作者还分享了7篇孟德尔随机化热点论文。

少数派网站发表的题为 “人工智能时代,重新发现骆驼 “的文章,讨论了以骆驼家族命名的人工智能模型的出现,骆驼家族是一个包括骆驼、羊驼、瓜纳科和骆驼在内的动物群体。文章首先介绍了骆驼如何成为中国流行的网络流行语的简要历史。然后,文章深入探讨了骆驼家族的生物分类,解释了各种物种之间的差异。文章的主要重点是开发以这些动物命名的人工智能模型。2023年2月,Meta AI发布了一个大型语言模型,名为LLaMA(大型语言模型Meta AI)。尽管LLaMA的参数比OpenAI的GPT-3少,但它的意义在于它是在非商业许可下发布的,主要用于学术研究。在LLaMA发布之后,其他几个以骆驼家族成员命名的AI模型也被开发出来。这些模型包括斯坦福大学基础模型研究中心开发的Alpaca 7B、Guanaco、来自不同大学的团队开发的Vicuna-13B,以及Chines-Vicuna。这些模型中的每一个都是通过用特定的数据集对LLaMA进行微调而开发的。文章最后指出了这些人工智能模型的快速发展,并将其比作新生骆驼的快速成熟,它在出生后就能立即奔跑。作者表示,希望在不久的将来,人工智能将变得人人可及。
8、Why it’s worth making computational methods easy to use

约翰霍普金斯大学生物医学工程系的助理教授Jean Fan在Nature上发表的一篇博文。文章讨论了Jean Fan和她的团队发起的一个数字活动,该活动利用YouTube、GitHub和博客等平台,使每个人都能获得计算生物学工具。文章探讨了这样一个观点:一个计算方法,就像艺术一样,在被他人分享和使用之前是不完整的。这篇文章是《自然》职业社区的一部分,《自然》读者可以在这个平台上分享他们的职业经验和建议。
9、What makes a good computational genomics method?

多伦多大学的助理教授Kieran R Campbell的博文,讨论了创建和评估新的计算生物学方法的原则。该文章强调了从问题而不是解决方案开始的重要性,寻求新的视角而不是改进的分数,有一个评估优先的方法,以及平衡什么是重要的和什么是可能的。
工具¶
10、Trinka
Trinka,一个用于学术和技术写作的语法和校正的人工智能 (AI)工具。
11、PyComplexHeatmap | 绘制复杂热图的Python包
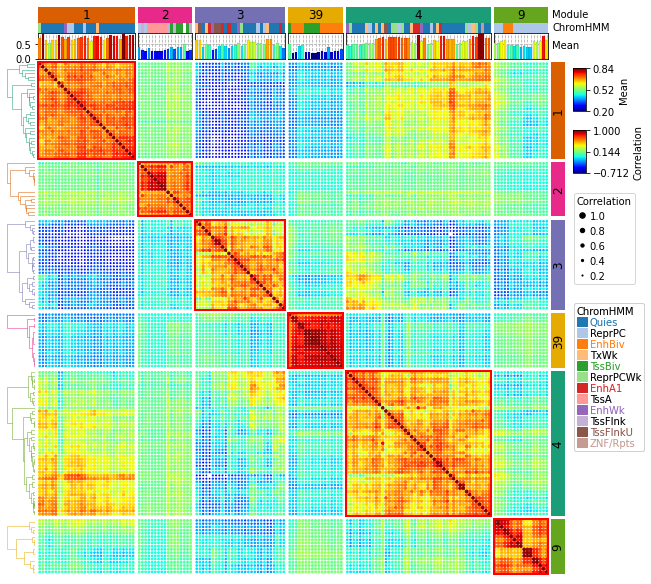
可以看作是 ComplexHeatmap 纯 Python 实现。我看了下,这不是 R 的接口调用。非常有意思的工作。
12、ChatPaper | 全流程加速科研:论文阅读+润色+优缺点分析与改进建议+审稿回复
ChatPaper是一款论文总结工具,AI用一分钟总结论文,用户用一分钟阅读AI总结的论文。
资源¶
13、计算机专业学习路线
本学习路线主要参考美国四大CS名校(卡耐基梅隆CMU、斯坦福Stanford、加州伯克利UC Berkeley、麻省理工MIT)以及哈佛Harvard、普林斯顿Princeton等6所计算机名校的课程安排与内容。而这六所名校也恰好是图灵奖得主最多的前六所,排名依次为:斯坦福、MIT、伯克利、普林斯顿、哈佛、CMU。世界上那些最优秀的IT名人可能都学过这些课程,耕耘其中,尽享奇妙。本学习路线会尽可能列出配套的视频、书籍、作业、项目等相关资源,而且会不断更新。
Ming Tommy Tang 在推特上总结的数据资料列表。
- RNA meta Analysis has ~26,700 studies (5,717 RNA-Seq and 20,955 Microarray) https://rnama.com/docs/search-evaluation
- refinebio will have harmonized over 60,000 gene expression experiments https://www.refine.bio/
- BioJupies https://maayanlab.cloud/biojupies/
- Recount2-FANTOM - Recounting the FANTOM Cage Associated Transcriptome. Long non-coding RNAs. https://www.biorxiv.org/content/10.1101/659490v1
- Recount3 https://rna.recount.bio/
- Digital Expression Explorer 2. Digital Expression Explorer 2 (DEE2) is a repository of uniformly processed RNA-seq data mined from public data obtained from NCBI Short Read Archive. https://dee2.io/
- Extracting allelic read counts from 250,000 human sequencing runs in Sequence Read Archive https://www.biorxiv.org/content/10.1101/386441v1?rss=1
- MetaSRA: normalized sample-specific metadata for the Sequence Read Archive https://www.biorxiv.org/content/10.1101/090506v1
- ARCHS4 provides access to gene counts from HiSeq 2000, HiSeq 2500 and NextSeq 500 platforms for human and mouse experiments from GEO and SRA. https://maayanlab.cloud/archs4/
- DEP-reads: Uniformlly processed public RNA-Seq data. Read counts data for 5,470 human and mouse datasets from ARCHS4 v6 and 12,670 datasets from DEE2 for 9 model organisms by steven Ge. http://bioinformatics.sdstate.edu/reads/
- SRA-explorer - This tool aims to make datasets within the Sequence Read Archive more accessible. https://sra-explorer.info/
- intropolis is a list of exon-exon junctions found across 21,504 human RNA-seq samples on the Sequence Read Archive (SRA) from spliced read alignment to hg19 with Rail-RNA. https://github.com/nellore/intropolis
- batch recompute ~20,000 RNA-seq samples from larget sequencing project such as TCGA, TARGET and GETEX. Used
hg38andgencode v21as annotation. https://toil.xenahubs.net - A cloud-based workflow to quantify transcript-expression levels in public cancer compendia used kallisto for TCGA/CCLE datasets and gencode v24 as annotation. https://www.biorxiv.org/content/10.1101/063552v1
- MiPanda is an online resource for the interrogation and visualization of gene expression data from the myriad of publicly available cancer and normal next generation sequencing datasets. https://mipanda.med.umich.edu
- Curation of over 10,000 transcriptomic studies to enable data reuse. https://biorxiv.org/content/10.1101/2020.07.13.201442v1
15、Single-cell RNA-seq data analysis workshop

一个单细胞分析入门教程的GitHub库。可以跟着助教学,也可以进行自学。教程提供了详细的数据和代码,值得花时间学学。
历史上的本周¶
- 第 39 期:人生不短
贡献者(GitHub ID)¶
「Openbiox 生信周刊」运维小队:
@ShixiangWang(王诗翔)@kkjtmac(阚科佳)@NiEntropy(赵启祥)@He-Kai-fly(何凯)@JnanZhang(张佳楠)@Tomcxf(陈啸枫)@wangdepin(王德品)@kongjianyang(空间阳)
订阅¶
这个周刊每周日发布,同步更新在微信公众号「生信协作组」(elegant-r)上。
微信搜索“生信协作组”或者扫描二维码,即可订阅。

(完)