生信爱好者周刊(第 52 期):真正的“科技与狠活”:全球首个人工“优选基因”的“完美婴儿”马上2岁啦!¶
这里记录每周值得分享的生信相关内容,周日发布。
本杂志开源(GitHub: openbiox/weekly),欢迎提交 issue,投稿或推荐生信相关内容。
封面图¶

位于 Loch Broom 岸边的Ullapool位于苏格兰北部高地的边缘。在这个崎岖的国家展现出它凄凉和孤独的辉煌之前,这是文明真正的最后一点。
本周话题:真正的“科技与狠活”:全球首个人工“优选基因”的“完美婴儿”马上2岁啦!¶

当科幻照进现实,人类基因组计划解码了人类基因,或能通过人类基因评估患病风险。走在科技前沿的科学家,总是第一个吃螃蟹的人。拥有遗传学博士学位的神经学家Rafal M Smigrodzki是全球首个为自己的女儿Aurea Yenmai Smigrodzki“优选基因”的父亲,被其称为“完美婴儿”的Aurea也即将健康成长至2周岁。”优选基因”带来降低疾病风险的优点的同时,也伴随着不断的伦理争议。
@ShixiangWang:科技从来都是双刃剑,我们喜欢它带给我们更多舒适精彩的生活,但又同时害怕它们带来的伤害和道德突破。在“优选基因”这个话题中,我觉得止不如疏,怎么联合起来设定科学且能接收的研究规范可能是未来需要讨论和解决的问题。
生信研究¶
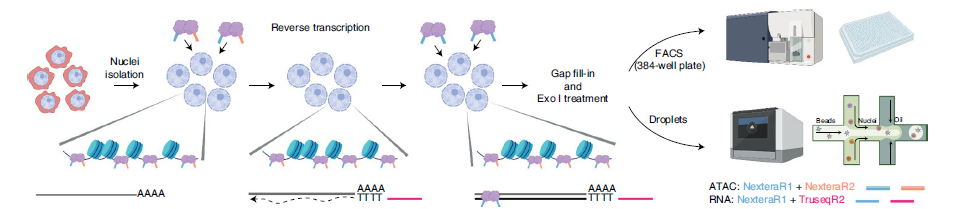
2022年9月15日,南方科技大学生命科学学院陈曦课题组和靳文菲课题组合作在Nature Methods上发表了一种简单可靠的单细胞多组学技术ISSAAC-seq,该方法能够在同一细胞内同时检测染色质可及性(ATAC-seq)以及基因表达(RNA-seq)。该方法操作灵活,既适用于基于孔板的低通量实验方案,又可以通过微流控装置进行高通量研究。
-
论文链接:https://www.nature.com/articles/s41592-022-01601-4

CITE-seq 分析的一个挑战是多个 CITE-seq 数据集的集成。这并非微不足道,因为不同 CITE-seq 数据集的蛋白质面板通常有一些不重叠情况,这会阻止简单的连接。为了规避这一点,研究人员为 sciPENN 引入了一种审查损失函数的方案,其中蛋白质损失被掩盖,并且当它从细胞中丢失时不会有助于反向传播。
这允许 sciPENN 从具有部分不重叠蛋白质面板的多个 CITE-seq 数据集中学习,估算组成每个 CITE-seq 数据集的缺失蛋白质,甚至从部分重叠的 CITE-seq 数据集学习后可以预测外部 scRNA-seq 数据集中的蛋白质的表达,这是 totalVI 和 Seurat 4 无法完成的。此外,sciPENN 比上述两个方法快一个数量级,使其成为综合 CITE-seq 和 scRNA-seq 数据分析的理想工具。
-
论文链接:https://www.nature.com/articles/s42256-022-00545-w

华盛顿大学的研究人员报告了一种基于深度学习的估计器,称其为 Deep-SMOLM,它在理论极限的 3% 内实现了 3D 方向和 2D 位置的卓越的测量精度(3.8° 方向、0.32 sr 摆动角和 8.5 nm 横向位置,使用 1000 个检测到的光子)。
-
论文链接:https://pubmed.ncbi.nlm.nih.gov/36258598/
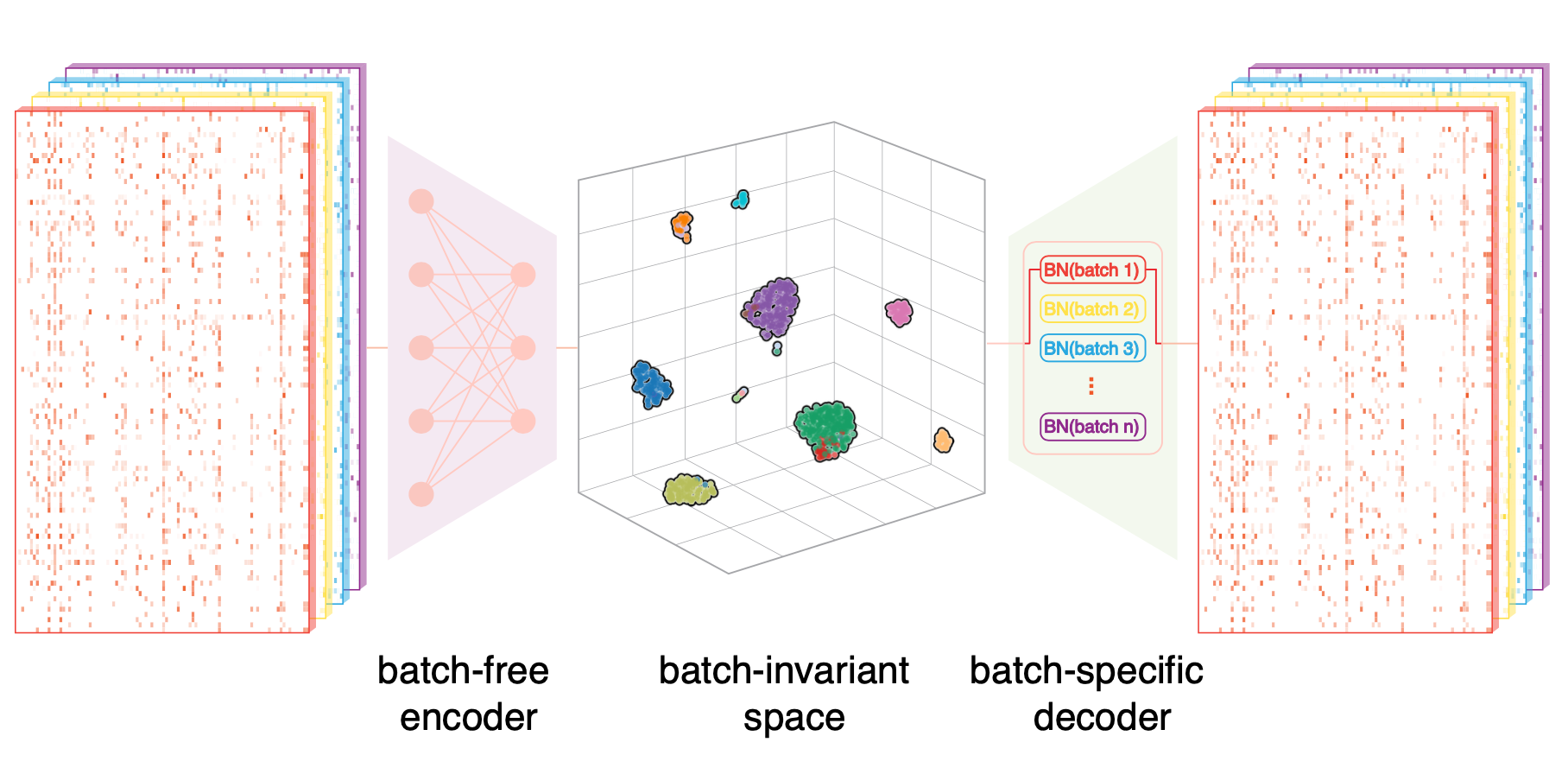
SCALEX是一款基于深度学习的单细胞整合算法。其通过将细胞投射到批次不变的、共同的细胞嵌入空间,即每次投影整合都不需要重新训练模型的方法,节省了计算成本。另外,在线数据整合能力和卓越的性能也使SCALEX特别适合于大规模的单细胞应用。 - Github:https://github.com/jsxlei/SCALEX - 论文链接:https://www.nature.com/articles/s41467-022-33758-z
博文资讯¶

近日,美国杰克逊基因组医学实验室的Francesca Menghi 和Edison T.Liu在Nature Communications总结分析了在癌症功能基因组学领域,科学家们是如何利用新一代测序技术、功能基因组学方法(通常与系统生物学方法相结合)揭示新的癌症发病机制。并提出这些最新成果超越了早期“一种基因、一种表型”的原始研究范式。
-
论文链接:https://www.nature.com/articles/s41467-022-33717-8

本文从Dockerfile出发,详细介绍了Docker 镜像构建中的具体参数和指令。
该推文分享了鄂维南院士在2022年的国际数学家大会对机器学习数学本质的理解,即函数逼近、概率分布的逼近与采样、Bellman方程的求解、机器学习模型的逼近误差、泛化性质以及训练等方面的数学理论,以及如何利用机器学习来求解困难的科学计算和科学问题,即AI for science。
-
视频:再谈AI for Science:https://www.bilibili.com/video/BV1vR4y1c7gQ/?vd_source=3afc2da90da868f4f01d17b6a37ffeba

本文从定义、原理、分子功能以及生物信息学中的应用四大方面,介绍了相分离这一物理化学概念。
工具¶

UCell是一个可以根据自定义的基因集给单细胞进行打分的工具,打分对象可以是矩阵、SingleCellExperiment或者Seurt对象。
-
论文链接:https://www.sciencedirect.com/science/article/pii/S2001037021002816?via%3Dihub

sc-linker是一个可以整合scRNA-seq、表观基因组单核苷酸多态性(SNP)- 基因图谱和GWAS统计数据的计算框架,并能够识别遗传变异影响疾病的潜在细胞类型和过程。
- 代码仓库:https://github.com/karthikj89/scgenetics
-
论文链接:https://doi.org/10.1038/s41588-022-01187-9

Sangerbox (http://vip.sangerbox.com) 是一个基于网络的工具平台,用户可以在其提供的友好的交互页面中进行不同分析。平台提供一系列可交互的图形化分析工具,包括相关性、通路富集、WGCNA分析等常见的工具和功能。
- 论文链接:https://onlinelibrary.wiley.com/doi/full/10.1002/imt2.36
资源¶

本推文整理了数据科学项目,包括数据导入、整理以及转换等过程所常用的R包。

该库收录了一系列Shiny相关的教程、工具、软件包、图书、论坛。学习Shiny,值得收藏。
-
仓库链接: https://github.com/grabear/awesome-rshiny
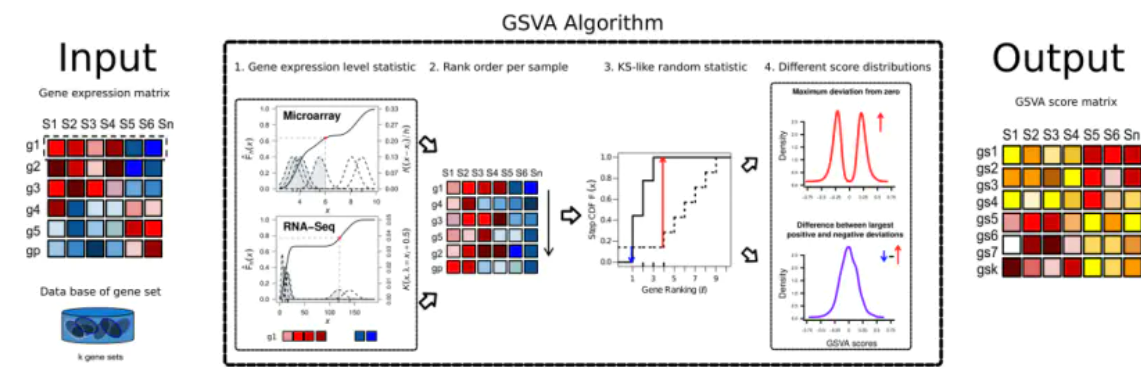
基因集变异分析(GSVA)是一种特殊类型的基因集富集方法,它可以对单个样本进行分析,并通过在基因到基因集的分析功能单元中执行概念上简单但功能强大的变化,实现以通路为中心的分子数据分析。
GSVA包提供了四种单样本基因集富集的方法,具体为zscore、plage、ssGSEA和它自己的GSVA。虽然它们最初是为基因表达数据开发的,但也可以应用于其他类型的分子分析数据。本教程中我们演示了如何使用GSVA包处理microarray和RNA-seq表达数据。
历史上的本周¶
贡献者(GitHub ID)¶
「Openbiox 生信周刊」运维小队:
@ShixiangWang(王诗翔)@kkjtmac(阚科佳)@NiEntropy(赵启祥)@He-Kai-fly(何凯)@JnanZhang(张佳楠)@Tomcxf(陈啸枫)@wangdepin(王德品)
订阅¶
这个周刊每周日发布,同步更新在微信公众号「生信协作组」(elegant-r)上。
微信搜索“生信协作组”或者扫描二维码,即可订阅。

(完)