生信爱好者周刊(第 44 期):为何动物的寿命差异那么大?¶
这里记录每周值得分享的生信相关内容,周日发布。
本杂志开源(GitHub: openbiox/weekly),欢迎提交 issue,投稿或推荐生信相关内容。
封面图¶

本周话题:为何动物的寿命差异那么大?¶
本周话题来自《测序中国:为何动物的寿命差异那么大?》,对动物寿命差异感兴趣的可以详细阅读下。

在哺乳动物体内,体细胞的突变会在健康细胞中终生积累。这些突变是肿瘤发生的基础,并被推测是导致衰老的原因之一。近日,英国Sanger研究所等单位的联合研究团队在Nature发表了题为“Somatic mutation rates scale with lifespan across mammals”的研究文章。研究表明,所有物种的体细胞突变主要由内源性突变过程主导,但体细胞突变率在不同物种之间差异较大,且与物种寿命呈强烈的负相关。总之,该研究数据揭示了哺乳动物的共同突变过程,并表明体细胞突变率在进化上受限,也可能是衰老的一个重要贡献因素。
@He-Kai-fly - 动物的寿命都是自然进化的结果,千百年来慢慢形成的,不同动物寿命不同,是因为养育后代所需要的时间以及自身所能承受的代谢结果对环境的适应程度的不同所导致的。
生信研究¶
1、Nature Machine Intelligence| 上交大团队开发预测人类白细胞抗原结合的 Transformer 框架
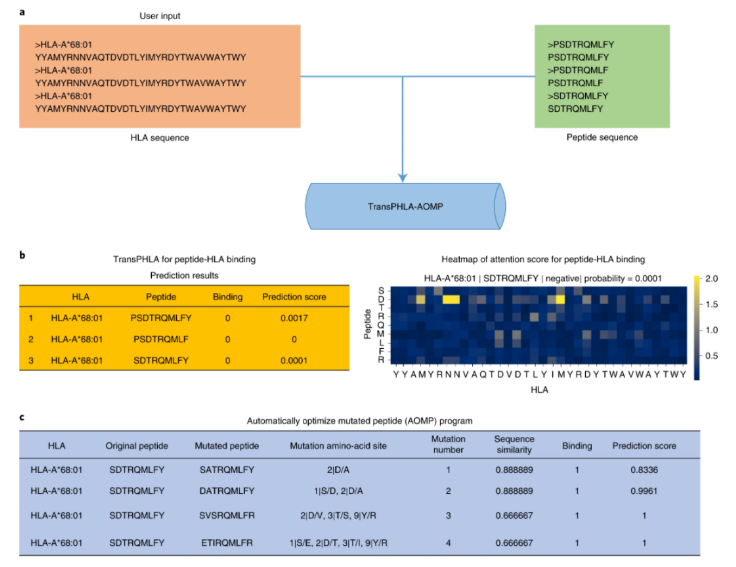
人类白细胞抗原(HLA)可以识别并结合外源肽,将它们呈递给专门的免疫细胞,然后启动免疫反应。肽和 HLA 结合的计算预测可以加速免疫原性肽筛选并促进疫苗设计。然而目前缺乏一个自动程序来优化对目标 HLA 等位基因具有更高亲和力的突变肽。为了填补这一空白,上海交通大学的研究人员开发了 TransMut 框架——由用于 pHLA 结合预测的 TransPHLA 和一个自动优化的突变肽 (AOMP) 程序组成——它可以推广到生物分子的任何结合和突变任务。该框架可应用于任何生物分子突变任务,例如表位优化或药物设计,尤其适用于疫苗开发。
论文链接:https://www.nature.com/articles/s42256-022-00459-7
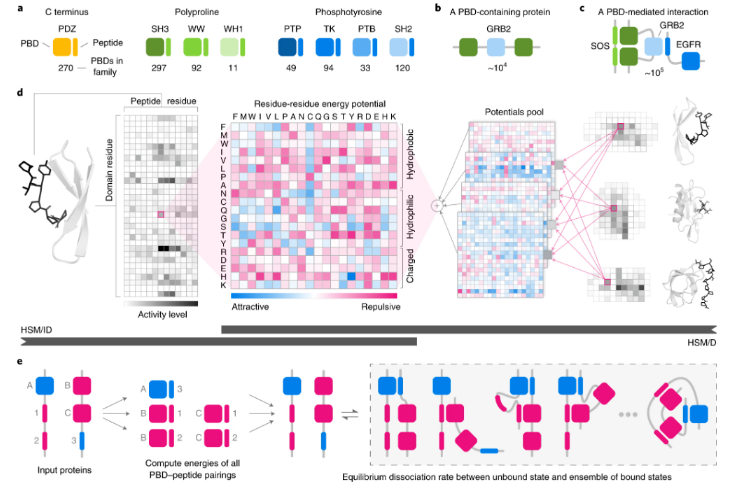
介绍四篇发表在【Nature Methods】上的高质量文章,分别提出了: - 剪接Z评分spliZ的统计方法、用于检测单细胞RNA测序中的调节剪接。 - 用于量化sc/snRNA-seq数据的alevin-fry框架 - 结合自编码期和潜在空间向量算法的高维单细胞及表达数据的模型—scGen - 一个定制的分层统计力学建模HSM,用于预测多个蛋白质家族PBD-肽相互作用的亲和力。
论文对应链接: 1. https://www.nature.com/articles/s41592-022-01400-x 2. https://www.nature.com/articles/s41592-022-01408-3 3. https://www.nature.com/articles/s41592-019-0494-8 4. https://www.nature.com/articles/s41592-019-0687-1
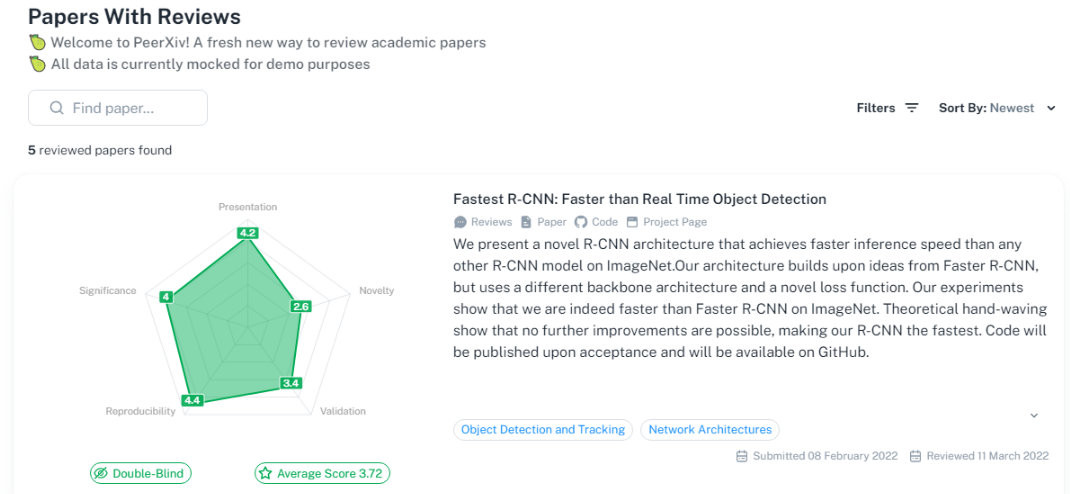
高效、透明的同行评审新平台PeerXiv ,论文作者提交预印本,5 位审稿人被要求按照 5 分制、5 指标的评价系统(新颖性、重要性、复现性、验证性、展现性)对每篇论文进行打分,论文作者一个月内就可收到审稿结果。
网址:https://peerxiv.web.app/dashboard/papers
4、DrugCVar|一个针对特定变异位点的肿瘤靶向用药分析平台
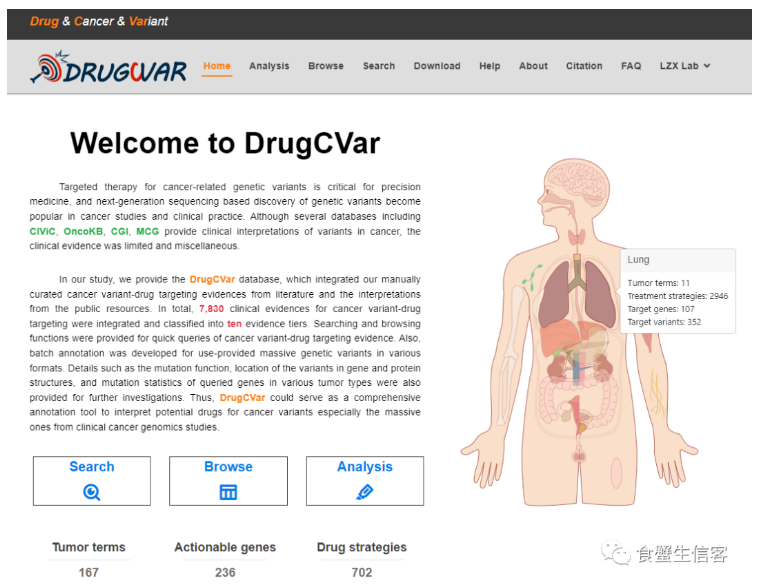
DrugCVar是由中山大学肿瘤防治中心刘泽先研究员团队开发的一个针对特定变异位点的肿瘤靶向用药分析平台。该平台整合了近几年发表的临床试验文献以及公开数据库 (OncoKB、CIViC、CGI和MCG) 的数据方便用户快速检索肿瘤变异靶向用药方案,同时对不同格式的肿瘤变异位点数据进行批量注释,为研究肿瘤的潜在靶向突变和制定治疗策略提供了重要数据支持。
论文链接:https://academic.oup.com/bioinformatics/article-abstract/38/11/3094/6569075?redirectedFrom=fulltext
博文资讯¶

这个帖子分享了许多研究生常逛的网站工具,建议新入学的研究生们看看。

本推文介绍了两种获取基因有效长度的方法,一是从上游输出文件结果中获取,二是从gtf文件中计算获取,同时附上了相应的代码和注释。
 介绍两种常用的查看R-package中某个函数的源代码方式,更加有效的学习优秀的R包提高对代码的理解能力。
介绍两种常用的查看R-package中某个函数的源代码方式,更加有效的学习优秀的R包提高对代码的理解能力。
链接: 1. https://rdrr.io/ 2. https://stackoverflow.com/questions/19226816/how-can-i-view-the-source-code-for-a-function
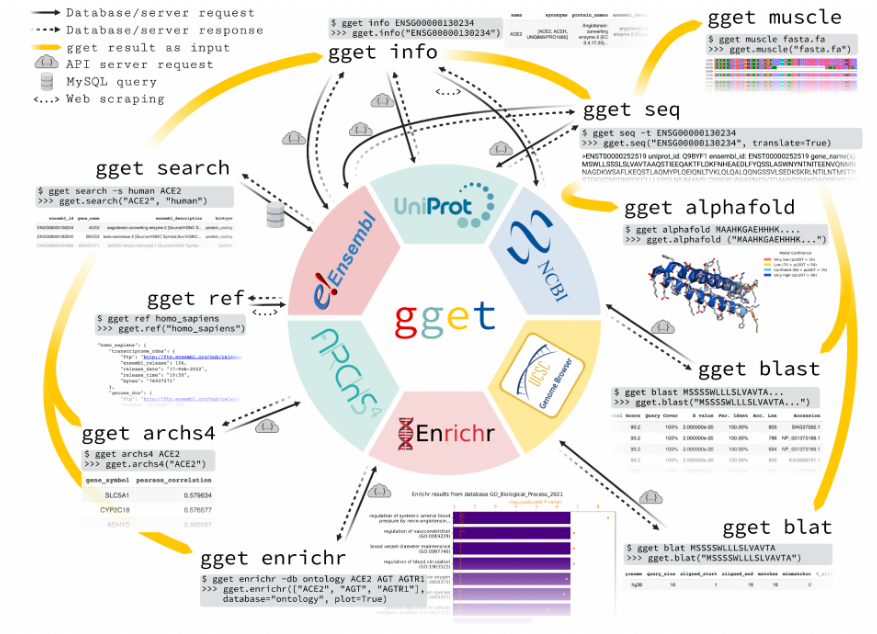
gget 软件包可以帮助我们直接从命令行或 Python 环境中快速查询存储在几个大型公共数据库中的信息。gget 由九个小工具组成,除了提供对基因组数据库的访问,还包括了一些分析工具,如 BLAST,简化了复杂的注释流程。每个 gget 工具仅需要很少的参数,就可以提供清晰完整的输出,最大程度地提高了易用性,对新手较为友好。
论文链接:Luebbert, L. & Pachter, L. (2022). Efficient querying of genomic reference databases with gget. bioRxiv 2022.05.17.492392;
doi: https://doi.org/10.1101/2022.05.17.492392
工具¶
thematic包简化了 ggplot2、lattice和 base的 R 图形的主题设置,提供了多种主题。
R包链接:https://github.com/rstudio/thematic
10、miloR|基于KNN图对单细胞数据集进行丰度差异分析
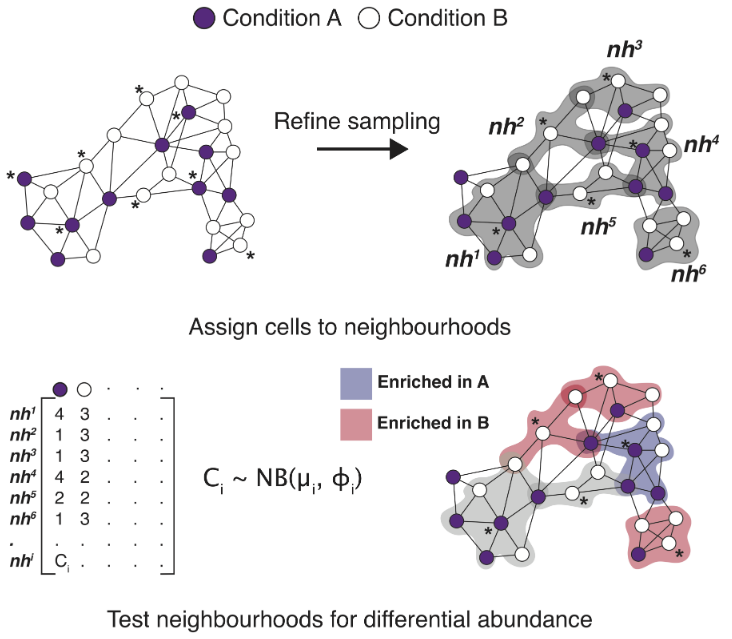
Milo是一种基于KNN图对单细胞数据集进行丰度差异分析的方法。
gitdown包可以用来创建gitbook。
 EasyStats是一系列包装,在使用R编程语言中的统计模型(R Core Team,2021)时,可在协同作用下运行,以提供一致和直观的语法。大多数EasyStats软件包返回模型参数和性能的全面数字摘要。See软件包通过许多功能和工具来补充这些数字摘要,以生成一系列可用于模型参数,预测和性能诊断的出版物的可视化。作为EasyStats的核心支柱,See软件包可帮助用户利用可视化,以提供更多信息,可传教和全面的科学报告。
EasyStats是一系列包装,在使用R编程语言中的统计模型(R Core Team,2021)时,可在协同作用下运行,以提供一致和直观的语法。大多数EasyStats软件包返回模型参数和性能的全面数字摘要。See软件包通过许多功能和工具来补充这些数字摘要,以生成一系列可用于模型参数,预测和性能诊断的出版物的可视化。作为EasyStats的核心支柱,See软件包可帮助用户利用可视化,以提供更多信息,可传教和全面的科学报告。
资源¶
13、shinymanager|为 Shiny 提供了登陆认证
 shinymanager 为 Shiny 提供了登陆认证的实现。
shinymanager 为 Shiny 提供了登陆认证的实现。
14、DGE_workshop|关于差异基因的Workshop资源

本资源是关于差异基因(DEG)分析的学习资料,从分析理论、所用R包和实例数据进行教学展示。
链接:https://hbctraining.github.io/DGE_workshop_salmon/schedule/
15、oncoEnrichR|对癌症背景下的人类基因组进行功能查询
oncoEnrichR是一个R软件包,用于对癌症背景下的人类基因组进行功能查询。它主要用于长基因列表的探索性分析、解释和优先排序。
历史上的本周¶
- 2021:第四期:生信有一天可以得诺贝尔奖吗
贡献者(GitHub ID)¶
「Openbiox 生信周刊」运维小队:
@ShixiangWang@kkjtmac@NiEntropy@He-Kai-fly@JnanZhang@Tomcxf@wangdepin
订阅¶
这个周刊每周日发布,同步更新在微信公众号「生信协作组」(elegant-r)上。
微信搜索“生信协作组”或者扫描二维码,即可订阅。

(完)